兰索拉唑肠溶微丸的制备和质量研究
2011-11-20陈志忠廉洁郝继红郝婷
陈志忠 廉洁 郝继红 郝婷
(1.内蒙古鄂尔多斯市药品检验所 内蒙古鄂尔多斯 017000; 2.北京悦康科创科技有限公司药学部 北京 100176;3.内蒙古鄂尔多斯市伊旗医院 内蒙古鄂尔多斯 017000)
兰索拉唑肠溶微丸的制备和质量研究
陈志忠1廉洁2郝继红1郝婷3
(1.内蒙古鄂尔多斯市药品检验所 内蒙古鄂尔多斯 017000; 2.北京悦康科创科技有限公司药学部 北京 100176;3.内蒙古鄂尔多斯市伊旗医院 内蒙古鄂尔多斯 017000)
以蔗糖、淀粉为稀释剂,以低取代羟丙基纤维素为崩解剂,碳酸氢钠为碱性添加剂以增加制剂的稳定性,以低粘度的羟丙基纤维素一定比例的乙醇溶液为粘合剂,并添加一定量的表面活性剂吐温-80,来增加主药兰索拉唑的释放量。通过离心造粒-包衣设备,采用粉末层积法制备兰索拉唑含药丸芯,然后分别包上隔离衣与肠溶衣,制得兰索拉唑肠溶微丸,再装入1号胶囊中,制得兰索拉唑肠溶胶囊。经过稳定性研究和耐酸力以及释放度检查,各项指标均符合要求。
兰索拉唑 肠溶微丸 离心造粒
兰索拉唑(Lansoprazole)[1]是继奥美拉唑之后由日本武田公司开发的世界上第二个质子泵抑制剂类抗溃疡药,1992年初,由武田公司和Houde公司在法国正式投放市场,1995年5月获FDA批准后在美国上市。兰索拉唑主要用于治疗胃溃疡、十二指肠溃疡、反流性食管炎、佐-艾(Zollinger-Ellison)综合征(胃泌素瘤)、吻合口部溃疡。天津武田公司生产的兰索拉唑肠溶胶囊,以商品名“达克普隆”在我国上市,台湾南光化学制药股份有限公司生产的兰索拉唑肠溶胶囊,商品名“拉索脱”,2005年在大陆上市销售,规格:30mg。
目前国内虽有兰索拉唑肠溶片剂上市,但因采用普通湿法制粒,生物利用度较低,且稳定性较差,并且,可能因意外因素致使肠溶衣破损,药物全部释放至局部药物浓度较高,有产生毒副作用的风险。本方法是将兰索拉唑和辅料混合后,采用离心造粒法制得含药丸芯,再分别包上隔离衣与肠溶衣,制得兰索拉唑肠溶微丸。服用本品后,会在肠道部位形成上百个小的释放单元,从而降低意外崩漏的风险,并大大提高了药品的生物利用度。
1 仪器与试药
UV-2401PC型紫外分光光度计,LC-10AD型高效液相色谱仪(日本岛津公司);RCZ-8G型智能溶出仪(天津大学无线电厂);Glatt流化床一步造粒机(上海格拉特公司)。兰索拉唑原料药(晋城海斯制药,批号:20090901)、兰索拉唑对照品(批号:100402,含量99.9%)均由药物研究所合成室提供;低取代羟丙纤维素(湖州展望化学药业有限公司,批号:2009012110),羟丙纤维素(日本曹达株式会社,批号:NOA-5511),淀粉(湖州展望化学药业有限公司,批号:20090408);蔗糖(上海运宏化工精细辅料技术有限公司代理,荷兰OMV公司生产,批号:10160993),隔离层包衣粉(胃溶型欧巴代)、肠溶包衣水分散体:EUDRAGIT L-30D-55,由赢创德固赛医药技术有限公司提供。
2 方法与结果
2.1 粉末层积法制备含药丸芯
2.1.1 制备丸芯 称取各辅料分别粉碎过160目筛,兰索拉唑粉碎过200目筛;取兰索拉唑150g、蔗糖150g、淀粉150g与碳酸氢钠150g,混合均匀。取1.0g吐温-803%(g/g)的羟丙纤维素30%乙醇溶液做为粘合剂。置离心-包衣造粒机中,转盘转速120~180r/min,供粉机转速15~40r/min,开启喷浆机,喷入上述粘合液,喷液速率为8.0g~20.0g/min,干燥进风温度为30~40℃,物料温度为25~35℃。
2.1.2 包隔离衣 筛分所制备的丸芯,收集20~30目之间的丸芯,以10%(g/g)胃溶型欧巴代70%乙醇溶液,进行隔离层包衣(增重14%)。
2.1.3 包肠溶衣 以德国赢创德固赛集团生产的EUDRAGIT系列产品:EUDRAGIT L-30D-55水分散体141g,柠檬酸三乙酯4.2g,滑石粉(1250目)12.5g,加水142g剪切15min后,包肠溶衣(增重22%~24%)。将制备的微丸,置38℃的恒温鼓风干燥箱内,处理120min,即得兰索拉唑肠溶微丸。
2.1.4 成品 采用1号胶囊灌装、检验、包装,即得兰索拉唑肠溶胶囊(规格:30mg/粒)。
依法制备本品三批样品各10000粒,批号为:100915、101019、101026。本品三批样品,均为白色肠溶球状小丸。
2.2 有关物质检查(避光操作)
取本品内容物,研细,称取细粉适量(约相当于兰索拉唑19mg),置50mL量瓶中,加稀释剂[水-乙腈-三乙胺(340∶160∶1),用磷酸调节pH值至10.0]适量,超声使兰索拉唑溶解并用稀释剂稀释至刻度,摇匀,离心,经滤膜(孔径不得>0.22μm)滤过,滤液作为供试品溶液;精密量取1mL,置100mL量瓶中,加稀释剂稀释至刻度,摇匀,作为对照溶液。照含量测定项下的色谱条件,以水-A液(60∶40)为流动相,理论板数按兰索拉唑峰计算不低于5000。取对照溶液20μL注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的10%~20%;再精密量取供试品溶液与对照溶液各20μL,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的3.5倍。供试品溶液的色谱图中如有杂质峰,2-[[[3-甲基-4-(2,2,2-三氟乙氧基)-2-吡啶基]-甲基]磺酰基]苯并咪唑(杂质A)按校正后的峰面积计算(乘以校正因子1.1),不得大于对照溶液主峰面积的0.5倍(0.5%),其他单个杂质峰面积不得大于对照溶液主峰面积的0.1倍(0.1%),杂质A乘以校正因子后与其他杂质峰面积的和不得大于对照溶液主峰面积(1.0%)。A液:乙腈-水-三乙胺(160∶40∶1),用磷酸调节pH值至7.0。

表1 三批样品有关物质检测结果(%)
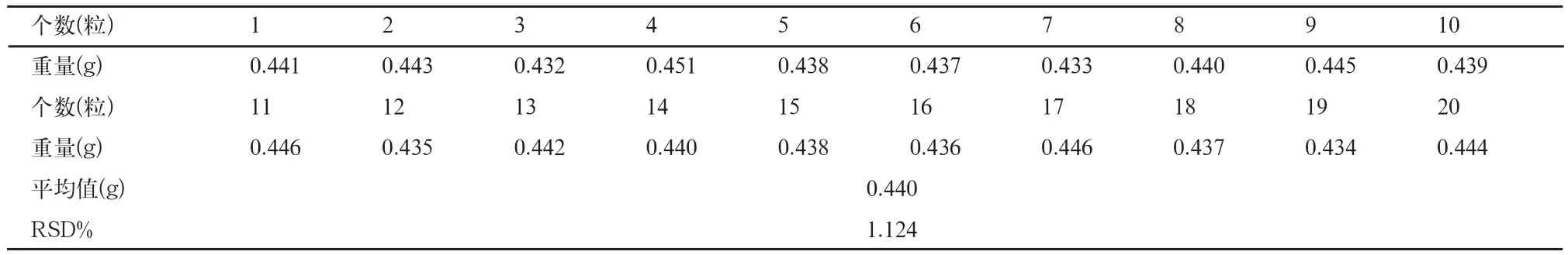
表2 装量差异检查结果
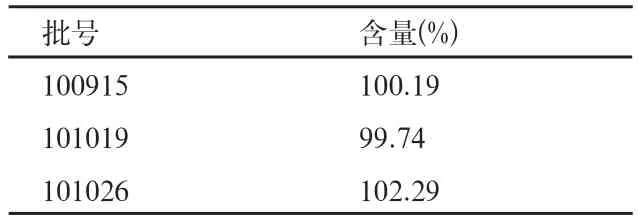
表3 含量检测结果
取本品样品三批,依法进行有关物质检查,结果见表1。
结果可见,三批样品的外观性状和有关物质均符合要求。
2.3 重量差异检查
取兰索拉唑肠溶胶囊20粒,照中国药典2010年版二部附录Ⅰ A重量差异方法检查,结果见表2。
结果表明,本品装量差异的RSD%<2%,装量符合要求。
2.4 含量测定
照高效液相色谱法(中国药典2010年版二部附录V D)测定。
色谱条件与系统适:用性试验用十八烷基硅烷键合硅胶为填充剂;以水-乙腈-三乙胺(60∶40∶1),用磷酸调节pH值至7.0为流动相;检测波长为285nm。理论板数按兰索拉唑峰计算不低于4000,兰索拉唑与内标物质峰的分离度应符合要求。
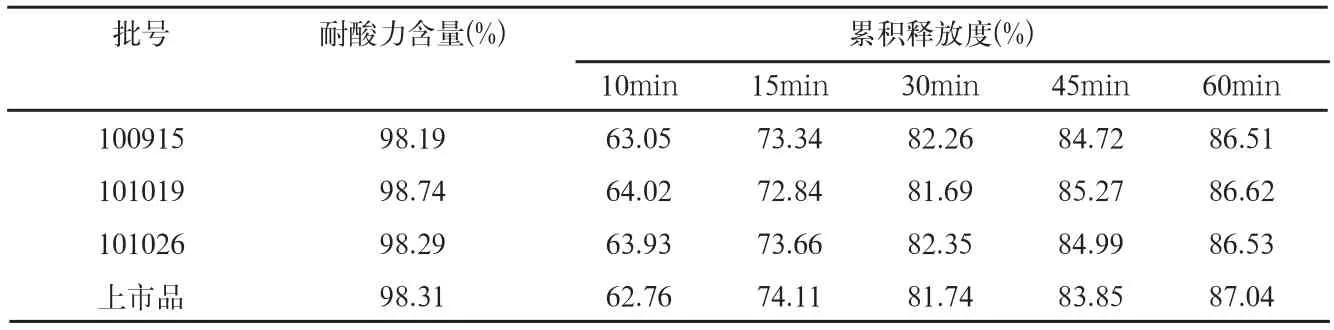
表4 耐酸力和释放度检测结果
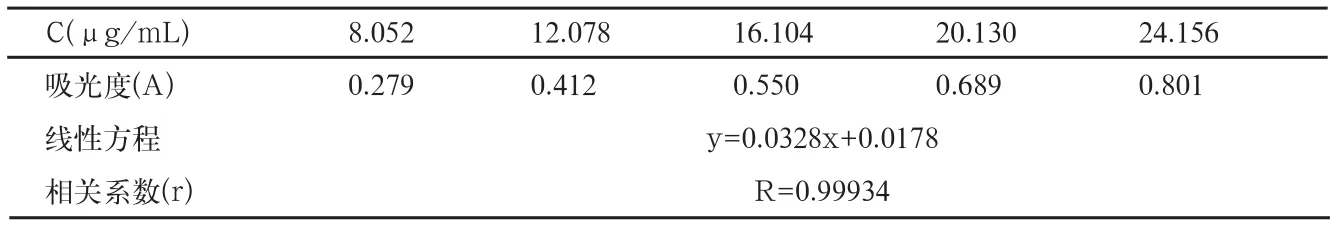
表5 线性与范围
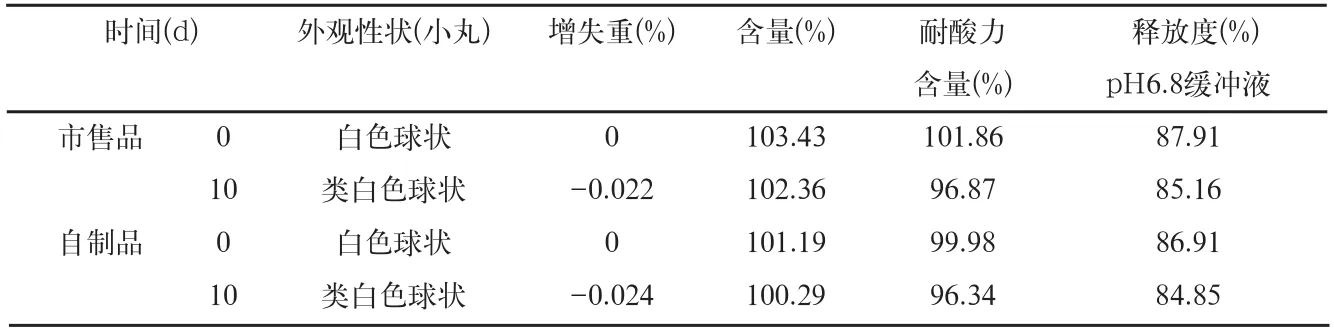
表6 影响因素0天和高温60℃检查结果
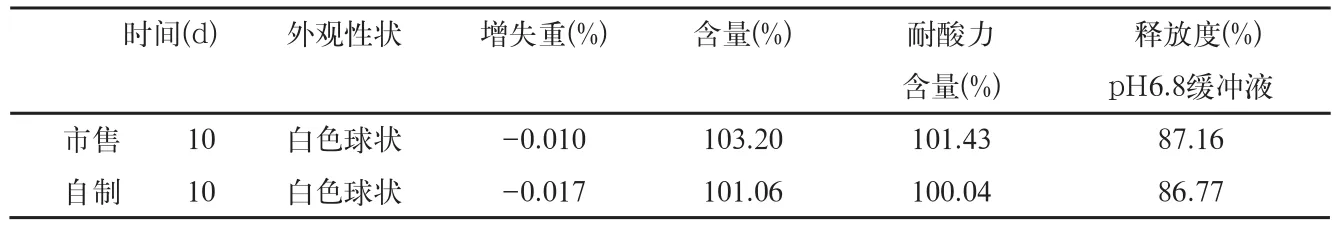
表7 影响因素高温40℃检查结果
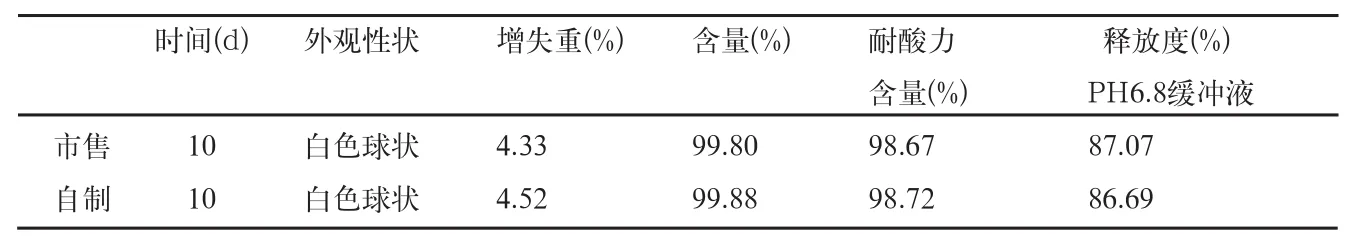
表8 影响因素高湿RH75%检查结果
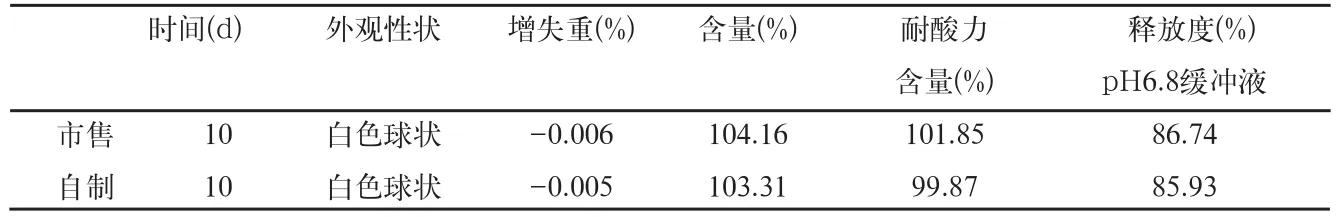
表9 影响因素光照检查结果
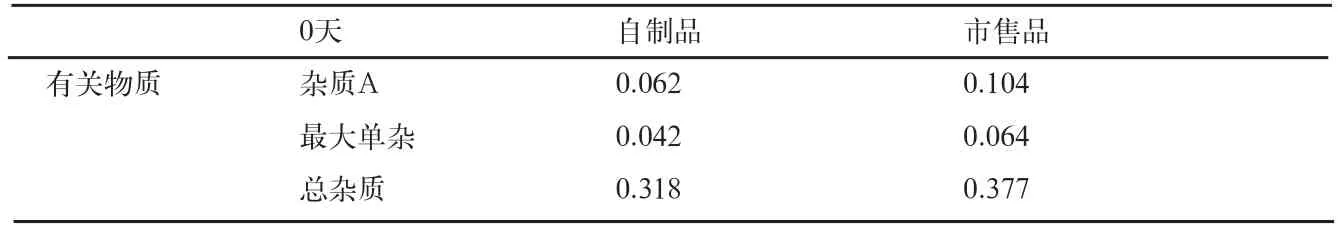
表10 影响因素0天有关物质检查结果(%)
内标溶液的制备:取4′-乙氧基乙酰苯,加乙腈制成每1mL中含5mg的溶液,即得。
测定法:取本品20粒,精密称定,将内容物混合均匀,研细,精密称取适量(约相当于兰索拉唑50mg),置50mL量瓶中,加0.1mol/L氢氧化钠溶液25mL,超声使兰索拉唑溶解,放冷,精密加内标溶液5mL,摇匀,用0.1mol/L氢氧化钠溶液稀释至刻度,摇匀,以3000转/分钟的速度离心5min,精密量取上清液5mL,置50mL量瓶中,加稀释剂[水-乙腈-三乙胺(60∶40∶1),用磷酸调节pH值至10.0]稀释至刻度,摇匀,滤过,精密量取续滤液20μl注入液相色谱仪,记录色谱图;另取兰索拉唑对照品约50mg,精密称定,置50mL量瓶中,加0.1mol/L氢氧化钠溶液25mL,超声使溶解,放冷,精密加内标溶液5mL,摇匀,用0.1mol/L氢氧化钠溶液稀释至刻度,摇匀,精密量取5mL,置50ml量瓶中,加稀释剂[水-乙腈-三乙胺(60∶40∶1),用磷酸调节pH值至10.0]稀释至刻度,摇匀,同法测定,按内标法以峰面积计算,即得。
取本品样品三批,依法进行含量检查,结果见表3。
结果可见,三批样品的含量均符合要求。
2.5 体外耐酸力和释放度检查
耐酸力:取本品,照溶出度测定法(中国药典2010年版二部附录Ⅹ C第一法),以氯化钠的盐酸溶液(取氯化钠2g,加盐酸7mL,加水溶解并稀释至1000mL,pH值为1.2)900mL为溶出介质,转速为150转/min,依法操作,经60min时,取下转篮,用水洗转篮内颗粒至洗液呈中性,用少量甲醇将颗粒移至50mL量瓶中,超声使兰索拉唑溶解,用甲醇稀释至刻度,摇匀,经0.45μm微孔滤膜滤过,精密量取续滤液2mL,置100mL量瓶中,加释放介质pH6.8磷酸盐缓冲液,稀释至刻度,摇匀,做为供试品溶液;另取兰索拉唑对照品约30mg,精密称定,置50mL量瓶中同法配制,作为对照品溶液。取上述2种溶液,照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A),在284nm的波长处测定吸光度,计算出每粒的含量,6粒中每粒含量不得少于标示量的90%;如有1~2粒小于标示量的90%,平均含量不得少于标示量的90%。
释放度:取本品,照释放度测定法(中国药典2010年版二部附录Ⅹ D第二法),采用溶出度测定法(中国药典2010年版二部附录Ⅹ C第一法)装置,以氯化钠的盐酸溶液(取氯化钠2g,加盐酸7mL,加水溶解并稀释至1000mL,pH值为1.2)900mL为释放介质,转速为150转/min,依法操作,经60min时,将上述操作的酸溶液弃去,在操作容器中加入预热至(37±0.5)℃pH6.8磷酸盐缓冲液[0.1mol/L盐酸溶液-0.2mol/L磷酸三钠溶液(3∶1)混合均匀,必要时用2.0mol/L盐酸溶液或2.0mol/L氢氧化钠溶液调节pH值至(6.8±0.05)]900mL为释放介质,转速不变,继续操作,经60min时,取溶液30mL,经0.45μm微孔滤膜滤过,弃去初滤液20mL,精密量取续滤液5mL,用pH6.8磷酸盐缓冲液稀释至10mL,摇匀,作为供试品溶液;另精密称取兰索拉唑对照品20mg,置25mL量瓶中,加甲醇溶解并稀释至刻度,摇匀,精密量取2mL,置100mL量瓶中,用pH6.8磷酸盐缓冲液稀释至刻度,摇匀,作为对照品溶液。取上述2种溶液,照紫外-可见分光光度法(中国药典2010年版二部附录Ⅳ A),在284nm的波长处分别测定吸光度,计算每粒的释放量,限度为标示量的75%,应符合规定。
取本品三批样品,依法进行耐酸力和释放度检查,结果见表4。
拟定释放度曲线如图1。
结果可见,本品三批样品的释放度曲线,与上市品释放度曲线有较好的相似性。
2.6 线性与范围
取兰索拉唑对照品约20mg,置25mL容量瓶中,加甲醇适量,振摇使溶解,加甲醇至刻度,摇匀,作为储备液,备用。精密量取储备液1.0、1.5、2.0、2.5和3.0mL,置100mL容量瓶中,加pH6.8磷酸盐缓冲溶液稀释至刻度,配制成不同浓度的对照品溶液。按照溶出度项下的方法测定,以样品浓度(C)为横坐标,以相应的峰面积(A)为纵坐标,进行线性回归,结果见表5(图2)。
结论:在本方法条件下,兰索拉唑溶液浓度在8.000~24.000μg/mL范围内,浓度与其吸光度呈良好的线性关系。
3 稳定性研究
参照中国药典2010年版二部附录《药物稳定性试验指导原则》中影响因素实验方法,取自制胶囊和天津武田公司生产的市售品,商品名:达克普隆®,除去外包装后,倾出内容物肠溶微丸,放置在高温(60℃和40℃),高湿(相对湿度RH92.5%和RH75%)、光照(4500±500)lx条件下于第10天取样,对其外观性状、释放度、耐酸力、有关物质和含量等进行考察。
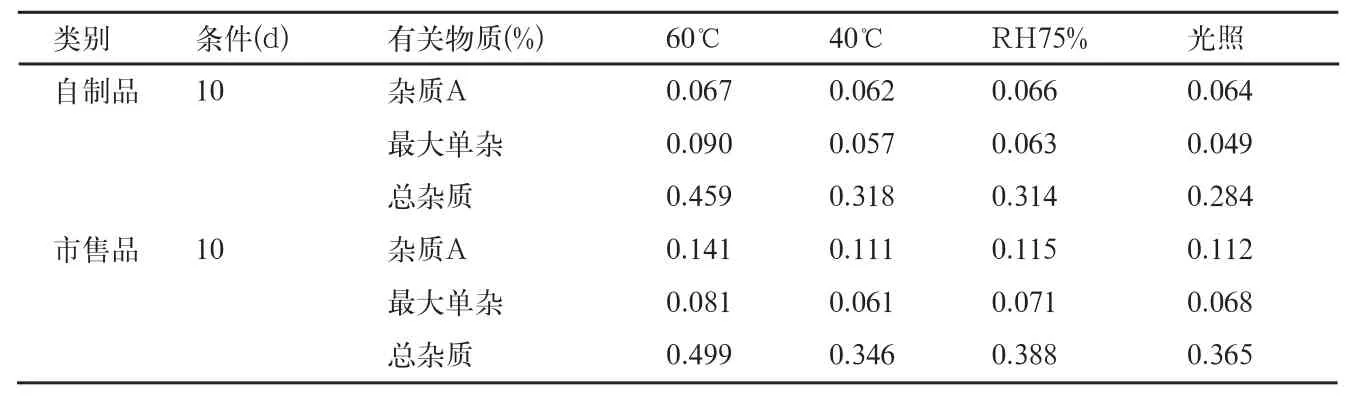
表11 影响因素5和10d有关物质检查结果
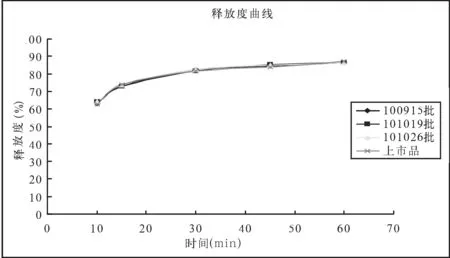
图1
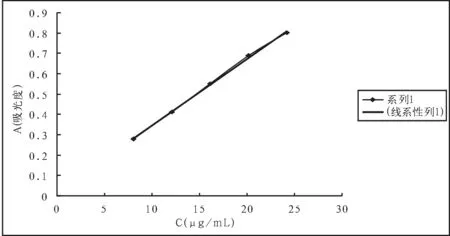
图2
3.1 高温试验
3.1.1 样品60℃条件检查结果(表6)本品在高温60℃条件下放置的样品,与零d的样品比较,放置10d的样品微丸颜色加深,呈类白色,耐酸力和释放度均略有下降。
3.1.2 样品40℃条件检查结果(表7)结果表明,本品在高温40℃条件下放置10d,与零天的样品比较,各项指标均无明显变化。
3.2 高湿试验
自制品和市售品肠溶小丸,放置在RH92.5%条件下5d后,均吸湿粘结成浆状物,故不做检查;放置在RH75%条件10d后,取样检查结果见表8。
结果表明,本品在RH75%条件下放置10d的样品,与零天的样品比较,各项检查无明显变化。
3.3 光照试验
取自制品和市售品肠溶微丸,置光照(4500±500)lx条件下10d后,取样检查,结果见表9。
结果表明,本品在光照(4500±500)lx条件下放置10d的样品,与零天的样品比较,各项指标均无明显变化。
3.4 有关物质检查结果
自制品和市售品肠溶微丸,在各个条件下放置10d后,有关物质检查结果见表10、11。
影响因素试验结论:本品高温(60、40℃)、高湿(RH92.5%、RH75%)及光照(4500±500)lx下,放置10d后,在高湿RH92.5%条件下,吸湿粘结成浆状物,故不再检测。高温60℃条件下,杂质A、最大单个杂质和总杂质均有不同程度的增加,但最大单个杂质<0.1%;其余各项指标均无显著性变化,自制品和上市品变化趋势一致。根据稳定性研究结果并参考上市品的包装说明,确定本品应密封,在阴凉(不超过20℃),干燥处保存。
4 讨论
(1)本品处方中以低取代羟丙基纤维素为崩解剂,低粘度的羟丙纤维素做粘合剂,蔗糖、淀粉做为稀释剂,能够制备流动性和坚固性良好的微丸,用吐温-80做为增溶剂,增加了兰索拉唑在pH6.8磷酸盐缓冲液中的释放量。以碳酸氢钠做为酸碱调节剂,并用欧巴代的醇溶液包裹了一定厚度的隔离层,较好的增加了本品的稳定性。
(2)由于兰索拉唑对光、热非常敏感,在生产与检验时应尽量避光操作。另外,本品微丸易吸潮,故在包隔离层时,需用欧巴代的醇溶液。使用EUDRAGIT L-30D-55水分散体包肠溶衣,不但可以节省设备成本,而且成膜性好,有较强的抗裂性和较好的韧性,包衣后微丸表面光洁美观。
[1]何小平.一种新的质子泵抑制剂-兰索拉唑[J].金陵医院学报,1996(2):160~162.
R2
A
1672-5654(2011)04(a)-0059-03
2011-03-28
