传统发酵豆制品中原核微生物多样性的研究
2011-11-02陈源源左志锐王正祥
陈 浩,樊 游,陈源源,左志锐,王正祥,*
(1.江南大学生物工程学院生物资源与生物能源研究中心,江苏无锡214122; 2.江南大学工业生物技术教育部重点实验室,江苏无锡214122)
传统发酵豆制品中原核微生物多样性的研究
陈 浩1,2,樊 游1,2,陈源源1,2,左志锐1,王正祥1,2,*
(1.江南大学生物工程学院生物资源与生物能源研究中心,江苏无锡214122; 2.江南大学工业生物技术教育部重点实验室,江苏无锡214122)
通过构建16S rDNA基因文库的方法,对2份不同种类的豆酱样品中原核微生物的多样性进行了分析。另外动态监测了一份酱油酱醅样品发酵过程中3个不同阶段原核微生物的变化情况。研究表明,发酵样品中的优势乳酸菌为魏斯氏菌(Weissella cibaria,Weissella confusa,Weissella paramesenteroides)和嗜盐四联球菌(Tetragenococcus halophilus)。另外还检测到芽孢杆菌、葡萄球菌、肠杆菌、柠檬酸杆菌、泛菌、不动杆菌、库特氏菌等细菌种群的存在。
传统发酵豆制品,16S rDNA,微生物多样性
1 材料与方法
1.1 材料与仪器
传统发酵黄豆酱、蚕豆酱各一份 采自安徽淮南;酱油酱醅三份 分别为发酵8、16、25d的样品,采自江苏无锡调味食品有限公司;培养携带T载体的Escherichia coli JM 109 含有100μg/mL氨苄青霉素的LB培养基;pMD 18-T载体 宝生物工程(大连)有限公司;质粒提取、DNA纯化试剂盒 QIAGEN公司;其他试剂 均为国产分析纯。

表1 2种不同豆酱中的细菌菌群分布
遗传分析仪Applied Biosystems 3130。
1.2 实验方法
1.2.1 样品总DNA的提取[5]取5g发酵样品,加入5mL DNA抽提缓冲液100mmol/L Tris-HCl pH8.0,100mmol/L EDTA pH8.0,100mmol/L磷酸盐缓冲液pH8.0,1.5mol/L NaCl,1%CTAB,研磨,再加入5mL DNA抽提缓冲液,加入50μL蛋白酶 K 20mg/mL,37℃恒温水浴1h,加入10%SDS 10mL,65℃恒温水浴2h,每隔10~20min轻轻摇匀,溶液加到离心管中,8000r/min离心10min,取上清液加至一新管中,加入等体积的氯仿混匀,8000r/min离心10min,取上清液至已预冷的新管中,加入0.6倍体积预冷的异丙醇,-20℃保存20min,8000r/min离心15min,弃上清,用70%的乙醇洗涤沉淀,晾干后溶于去离子水中,RNase消化后进行琼脂糖电泳检测。
1.2.2 16S rDNA基因文库构建 以提取的总DNA为模板,以原核生物通用引物 27F(5'-AGAGTTTGATCCTGGCTCAG-3')和 1492R(5'-ACGGCTACCTTGTTACGACTT-3')为上下游引物扩增16S rDNA基因。PCR条件为:94℃预变性5min; 94℃变性30s,52℃退火1min,72℃延伸90s,经过30个循环后,72℃再延伸10min。用0.8%(w/v)的琼脂糖凝胶电泳分离PCR产物。PCR产物经试剂盒纯化后连接pMD 18-T载体,热激转化Escherichia coli JM 109感受态细胞。以氨苄青霉素(100μg/mL)抗性和蓝白斑筛选来选择阳性转化子,并用质粒提取试剂盒提取质粒,通过电泳进一步验证16S rDNA的插入片段。
1.2.3 序列分析和系统发育树分析 阳性质粒转化子经 BigDyeTerminatorv3.1 试剂盒 (Applied Biosystems)进行测序,测序上下游引物为M13-47 (5'-CGCCAGGGTTTTCCCAGTCACGAC-3')和M13-48(5'-AGCGGATAACAATTTCACACAGGA-3')。测序后结果经过Sequence Scanner v1.0软件分析后提交至NCBI-BLAST进行序列同源性分析,选择相似性较高的相关菌株的基因序列,用ClustalW软件进行序列多重比对[6],用MEGA 4.0软件包采用邻接法(Neighbor-Joining)聚类分析和系统进化树构建[7]。重复取样1000次进行自展值分析以评估系统进化树的拓扑结构稳定性[8]。
2 结果与分析
2.1 豆酱样品中原核微生物的多样性
两种豆酱样品均取自安徽淮南,但是两种样品中的原核微生物却不尽相同。由表1可知,蚕豆酱中的乳酸菌为嗜盐四联球菌 (Tetragenococcus halophilus),而黄豆酱中的乳酸菌则以魏斯氏菌为主(Weissella cibaria和Weissella confusa),两种样品中乳酸菌分别占检测出细菌菌群的20%和12%。
蚕豆酱中检测到多种其他细菌,如肠杆菌(Enterobacter aerogenes,Enterobacter cancerogenus,Enterobacter sp.)、弗氏柠 檬 酸杆菌 (Citrobacter freundii)、成团泛菌(Pantoea agglomerans)、不动杆菌(Acinetobacter baylyi)等细菌种群都有被检测到。
而黄豆酱中的优势细菌种群则为芽孢杆菌属的细菌(Bacillus subtilis、Bacillus amyloliquefaciens),占检测总菌群的 72%,另外则有鸡葡萄球菌(Staphylococcus gallinarum)和肠杆菌(Enterobacter aerogenes)的存在。
我们发现两种相同产地的样品中所检测到的细菌菌群存在差异性,这可能与不同的制作原料和生产方法有关,也可能是发酵样品的不同pH所导致的。
2.2 酱油酱醅中原核微生物的多样性
通过对酱油发酵过程中的3个不同阶段的样品进行检测,动态跟踪了整个酱油发酵过程中的原核微生物的变化过程。总体来看,酱醅中的乳酸菌以魏斯氏菌(Weissella paramesenteroides和 Weissella confusa)为主。分阶段看,如图1所示,发酵8d的样品中,魏斯氏菌占61%,库特氏菌(Kurthia gibsonii)占11%,芽孢杆菌(Bacillus licheniformis)只占检测到细菌的6%。在发酵16d的样品中,魏斯氏菌降为42%,库特氏菌和芽孢杆菌则占比上升,分别达到25%。在这个阶段中,另有乳酸片球菌(Pediococcus acidilactici)被检测到。而在发酵25d的样品中,我们的研究只发现有芽孢杆菌的存在,而其他的细菌菌群都没有被检测到。
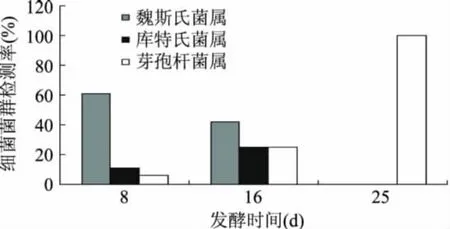
图1 不同发酵阶段酱醅中主要细菌菌群的变化
另外,在发酵8d的样品中,我们检测到的有22% 的克隆子分别与 Lactobacillus curvatu KH9 (AB494734)和 LactobacilluspontisLMG 14188 (AJ422033)的16S rDNA序列同源性达97%和95%,见图2。从构建的系统进化树上可以看出,检测到的克隆子(JP3和JP10)与其典型菌株以较高的自展值(92%和88%)支持聚在一个系统进化分支上,表明它们都是乳杆菌属(Lactobacillus)的一个种群。

图2 根据检测到的酱醅中原核微生物的16S rDNA序列构建的系统发育树
乳杆菌、片球菌是酱油发酵中的常见乳酸菌。而我们的实验却发现魏斯氏菌在某些发酵豆制品中占有优势地位。有报道称魏斯氏菌可以产生某些细菌素,抑制一些革兰氏阳性菌生长[9],这可能是在酱油发酵的前期芽孢杆菌较少的原因之一。另外,由于在发酵25d的时候,发酵温度上升至50℃左右,这也可能是最后只检测到芽孢杆菌存在的原因。
3 结论
本研究通过构建16S rDNA基因文库的方法,对2种不同种类的豆酱样品中的原核微生物进行多样性分析,并动态监控了酱油发酵过程中的原核微生物变化情况。研究结果表明,样品中的乳酸菌主要以魏斯氏菌和嗜盐四联球菌为主,另外也有芽孢杆菌和其他一些污染菌群存在。
利用构建基因文库的方法对自然发酵过程中微生物菌群构成进行分析,可真实、客观地反映微生物群落的结构和多样性,避免了传统分离方法的片面性,对人工发酵剂的设计以及传统发酵食品的工业化生产等方面的研究具有重要意义。
虽然构建基因文库的方法可以较好地反映出样品中微生物菌群的分布,但是采用这种方法对一个环境中样品多样性进行考察时,必须注意一些因素对研究结果造成的误差,如由于不同微生物的细胞壁结构差异引起的细胞裂解不完全,造成DNA获得的不均一性[10];编码16S rDNA的操纵子拷贝数的差异及不同DNA和PCR引物亲和力不同所引起的扩增产物浓度的差异[11]等。
[1]Golbitz P.Traditional soyfoods:processing and products[J]. Journal of Nutrition,1995,125(3):570-572.
[2]Park HK,Gil B,Kim JK.Characteristics of taste components ofcommercial soybean paste [J].Food Science and Biotechnology,2002,11(4):376-379.
[3]Hugenholtz P.Exploring prokaryotic diversity in the genomic era[J].Genome Biol,2002,3(2):1-8.
[4]Tyson GW,Chapman J,Hugenholtz P,et al.Community structure and metabolism through reconstruction of microbial genomes from the environment[J].Nature,2004,428:37-43.
[5]Zhou JZ,Bruns MA,Tiedje JM.DNA Recovery from Soils of Diverse Composition [J] .Applied and Environmental Microbiology,1996,62(2):316-322.
[6]Thompson JD,Gibson TJ,Plewniak F,et al.The CLUSTAL_X windows interface:flexible strategies formultiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Research,1997,25(24):4876-4882.
[7]Saitou N,Nei M.The neighbor-joining method:a new method for reconstructing phylogenetic trees[J].Molecular biology and evolution,1987,4(4):406-425.
[8]Felsenstein J.Confidence limits on phylogenies:an approach using the bootstrap[J].Evolution,1985,39(4):783-791.
[9]Srionnual S,Yanagida F,Lin LH,et al.Weissellicin 110,a Newly Discovered Bacteriocin from Weissella cibaria 110,Isolated from Plaa-Som,a Fermented Fish Product from Thailand[J]. Applied and Environmental Microbiology,2007,73(7):2247 -2250.
[10]Frostegard A,Courtois S,Ramisse V,et al.Quantification of bias related to theextraction of DNA directly from soils[J]. Applied and Environmental Microbiology,1999,65(12):5409 -5420.
[11] Martin-Laurent F,Philippot L,Hallet S,et al.DNA extraction from soils:old bias for new microbial diversity analysis methods[J].Applied and Environmental Microbiology,2001,67 (5):2354-2359.
Study on the prokaryotic microbial diversity of Chinese traditional fermented soybean foods
CHEN Hao1,2,FAN You1,2,CHEN Yuan-yuan1,2,ZUO Zhi-rui1,WANG Zheng-xiang1,2,*
(1.Center for Bioresource&Bioenergy,School of Biotechnology,Jiangnan University,Wuxi 214122,China; 2.The Key Laboratory of Industrial Biotechnology,Ministry of Education,Jiangnan University,Wuxi 214122,China)
The prokaryotic microbial diversities of two different soybean pastes were studied by the 16S rDNA gene libraries.In addition,the prokaryotic microbial community dynamics monitor of say sauce paste was reviewed.The results showed that Weissella cibaria,Weissella confuse,Weissella paramesenteroides and Tetragenococcus halophilus were the dominant lactic acid bacteria in samples.Bacillus,Staphylococcus,Enterobacter,Citrobacter,Pantoea,Acinetobacter and Kurthia were found in samples.
traditional fermented soybean foods;16S rDNA;microbial diversity
TS201.3
A
1002-0306(2011)09-0230-03
传统发酵豆制品在我国有悠久的生产历史,它们均是利用天然大豆加工而成,具有营养丰富、易于消化吸收等特点。发酵豆制品一般包括豆酱、酱油、豆豉、腐乳等食品[1]。它们的生产被认为是一个开放的、传统的过程,其中有多种微生物参与。在此过程中发生一系列复杂的生化反应,使其发酵制品具有特定的风味、口感和营养。发酵豆制品中的微生物组成对发酵豆制品的质量有重要影响[2]。由于参与发酵的微生物种类繁多,有的对发酵有利,有的对人体有害,所以分析发酵豆制品中微生物菌群的组成,揭示微生物种类和遗传的多样性,对于传统发酵食品的工业化生产、发酵调控、稳定及提高产品质量具有积极的指导意义。早期关于发酵豆制品中微生物多样性的报道基本是建立在可培养(culture-dependent)的方法之上的,这种方法仅局限于在特定培养基生长出来的微生物,而大部分的微生物却不能通过实验室的纯培养获得[3]。本研究利用构建16S rDNA基因文库方法,对2种不同种类豆酱样品中的原核微生物进行多样性分析,并全程监控了酱油发酵过程中的原核微生物变化情况。构建基因文库可以把单一的目的基因克隆到载体上,被检测出的微生物菌群可以精确到种的水平上,而且根据测序得到不同转化子的数量比例,可以揭示出样品中的优势微生物菌群[4]。
2010-09-19 *通讯联系人
陈浩(1985-),男,在读硕士,研究方向:微生物资源。
微生物资源国家级平台资助项目(2005DKA21208)。
