三元化合物ZnCrS2电子结构和半金属铁磁性的第一性原理研究*
2011-11-02程志梅王新强王风鲁丽娅刘高斌段壮芬聂招秀
程志梅 王新强王风 鲁丽娅刘高斌段壮芬聂招秀
三元化合物ZnCrS2电子结构和半金属铁磁性的第一性原理研究*
程志梅1)2)王新强1)王风3)鲁丽娅1)刘高斌1)段壮芬1)聂招秀1)
1)(重庆大学物理学院,重庆400044)
2)(重庆师范大学教育科学学院,重庆400047)
3)(重庆师范大学初等教育学院,重庆400700)
(2010年10月27日收到;2010年11月21日收到修改稿)
以闪锌矿相的ZnS 2×2×1超原胞为基础,通过将其中的Zn用Cr按1∶1配比进行了a和b两种不同位置的替换构造出了三元化合物ZnCrS2理论模型,然后采用基于密度泛函理论(DFT)的平面波超软赝势(PWPP)方法分别计算了两种不同模型ZnCrS2的电子结构和磁学性质.结果表明,两种模型的ZnCrS2的铁磁态都比反铁磁态更稳定,均是半金属铁磁体(半金属能隙分别为0.9631 eV和0.7556 eV),其中a位替换不但具有较大的半金属能隙,而且结构更稳定.文中对a位替换结构的自旋极化电子态密度、能带结构、磁矩等进行了详细分析.本文结果可望为实验研究提供指导和理论解释.
ZnCrS2,电子结构,半金属铁磁性,第一性原理
PACS:63.20.dk,75.50.Cc,76.50.+g,71.20.Lp
1.引言
自旋电子学是一门磁学和微电子学相交叉的前沿学科,它是利用电子的电荷与电子自旋来完成信息存贮与处理.利用自旋电子学制造出的电子器件将会推动信息科学等高科技领域的飞速发展,这种电子器件同传统电子器件比较,它具有非易失性[1]、数据处理速度快、功耗低以及集成度高等优点[2].目前自旋电子学材料研究最多的当属稀磁半导体(DMS)[3,4],因为它同时具有磁性和半导体特性,并且利用电子的电荷与电子自旋自由度,在理论和应用上都具有十分重要的意义.但其至今没能得到广泛的应用,原因之一是它的居里温度低于室温[5],另外是饱和磁化强度较低[6,7],加之这类材料的磁性原子的密度较小,这就使得最终器件的尺度不能太小.
半金属(half-metallic)铁磁体是自旋电子学材料的另一发展方向,它同时具有两种自旋行为:自旋向上的费米面穿过导带,表现出金属性,自旋向下的费米面处于导带和价带之间,表现出半导体性质.由于其有较高的居里温度和近100%的自旋极化率,半金属铁磁体是一种具有极大应用潜能的自旋电子学材料[8,9].
自1983年Groot等[8]率先发现合金NiMnSb和PtMn Sb具有半金属性后,人们相继从理论和实验中发现了铁磁性金属氧化物Sc M(M=C,Si,Ge,Sn)[10],CrO[11]2,Fe2O[12]3及二元闪锌矿(ZB)结构的过渡金属磷族化合物和硫族化合物CrSb[13],MnBi[14],CrAs[15,16],CrS[17]等具有半金属性,Li等人[18]还对原子无序性对full-Heusler合金Co2FeSi的半金属特性的影响进行了研究.
近年来的研究表明,在Ⅲ-Ⅴ族宽禁带半导体中掺入过渡金属同样具有半金属性,如Mn掺杂GaN[19],Cr,Mn掺杂AlN[20].基于Ⅱ-Ⅵ族半导体的三元化合物半金属铁磁体也逐渐被发现.王风等人[21]的计算表明,三元化合物ZnVSe2是一种半金属铁磁性材料;Fukumura等人[22]在蓝宝石衬底上利用脉冲激光淀积技术成功制备了ZnMnO;实验及理论计算表明Zn1-xMxTe(M=V,Cr;x=0.25,0.75)[23]也具有半金属性.然而对于Ⅱ-Ⅵ半导体中禁带最宽半导体的Zn S基的三元化合物半金属铁磁性的研究还鲜见报道.本工作是采用基于密度泛函理论(DFT)的平面波赝势方法(PWP),对基于ZnS的2×2×1超原胞通过替换构造出来的ZnCrS2八原子体系进行几何结构优化,研究它的电子结构和磁性,研究结果表明ZnCrS2为半金属铁磁体.
2.理论模型及计算方法
2.1.计算方法
本工作的计算是基于平面波基组的超软赝势从头算法,利用广义梯度近似(generalized gradient approximation,GGA)的PBE基组处理电子间的交换关联能.对替换后的ZnCrS2晶胞进行几何优化,为确保计算速度和计算精度,平面波截断能取420 eV,K点网格的大小为2×2×5,原子间相互作用收敛标准为0.01 eV/nm,单原子能量收敛标准为5.0 ×10-6eV/atm,原子最大位移收敛标准为0.0005,晶体内应力收敛标准为0.02 GPa,能量与性质计算时平面波截断能Ecut取为450 eV.参与计算的价态电子有S:3 s23 p4;Cr:3s23p63 d54 s2; Zn:3p63 d104s2.
2.2.理论模型
Zn S具有低温相立方闪锌矿(β-ZnS)和高温相六方纤锌矿(α-ZnS)两种晶体结构,当闪锌矿结构ZnS升温到1020℃时,即向六方纤锌矿结构转变.由于自然界中稳定存在的是闪锌矿结构,故本文计算采用的是立方闪锌矿结构的ZnS为基础构建三元化合物.为了研究不同位置替换对半金属带隙的影响,比较铁磁态和反铁磁态的能量高低,以判定体系的基态,计算时以ZnS原胞为基础,构建2×2×1超原胞,每个超晶胞中包含4个Zn原子和4个S原子,其晶格常数a=b=0.764991 nm,c=0.382495 nm,α=β=γ=60°.分别对超原胞不同位置的Zn原子用Cr原子进行替换,替换后所得三元化合物空间结构如图1所示.
按a位置(如图1(a)所示)替换后构建的三元化合物ZnCrS2的空间群为R3M(160),对称性为C2V-5,

图1 三元化合物ZnCrS2不同替换位置晶胞结构模型
优化前其晶格常数为a=b=0.764991 nm,c= 0.382495 nm,α=β=γ=60°,优化后其晶格常数为a= 0.783969 nm,b=0.766787 nm,c=0.382875 nm,α= 59.8815°,β=60.7470°,γ=60.8435°.按b位置(如图1(b)所示)替换后构建的三元化合物ZnCrS2的空间群为P-4 M2(115),对称性为D2 D-5,优化后其晶格常数为a=b=0.784726 nm,c=0.378911 nm,α=β=61.0065°,γ=57.7036°.
3.结果与讨论
3.1.ZnS的能带结构和态密度
为便于分析三元化合物Zn CrS2中Cr对ZnS体相电子结构等的影响,首先计算出了ZnS原胞的能带结构,结果如图2所示,相应的总态密度和分波态密度如图3所示.
从图2中可看出,纯Zn S晶体价带顶的导带底都位于布里渊区的G点,是直接能隙半导体.计算得出带隙值为2.23 eV,同实验值3.68 eV相比偏小.用密度泛函理论求解带隙时会比实验值偏小[24],这主要是由于理论本身过高地估计了Zn 3 d电子与S 3 p电子间的相互作用,导致价带宽度变大,带隙减小.从图3中可看出,ZnS的价带可分为两部分,0—-5.20 eV的上价带和-5.5—-6.4 eV的下价带.从图3可得出上价带主要由S 3p态贡献;下价带主要由高度局域的Zn 3d态贡献,还有极少量的S 3 p态.对于导带部分,主要来源于Zn 4s态和少量Zn 3 p的S 3 p态贡献.
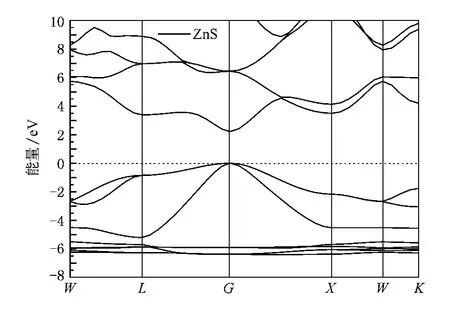
图2 ZnS能带结构

图3 ZnS态密度(a)S原子分态密度;(b)Zn原子分态密度; (c)ZnS总态密度
3.2.三元化合物ZnCrS2的能带结构和态密度
表1是a,b两种不同替换位置的计算结果,两种位置替换Zn CrS2的能带结构如图4和图5所示,电子态密度及分态密度的计算结果分别如图6与图7所示.
我们分别对Zn4S4进行了如图1(a),(b)两种不同位置的替换,两种不同位置替换后Zn CrS2的对称性分别为C2 V-5与D2 D-5,对两种不同位置替换的ZnCrS2优化后的晶格常数与优化前比较均有不同程度地增大,这主要是由于引入的Cr原子半径大于Zn原子所致.表1为不同替换位置计算结果,从表1中可知,a位置替换的铁磁态能量比反铁磁能量低0.155 eV;b位置替换的铁磁态能量比反铁磁能量低0.131 eV.显然,铁磁态是更稳定的基态.由表1还可得到a位置的铁磁态能量较b位置替换的能量低0.129 eV.故a位置较b位置更稳定.
如图4,图5所示,a,b两种位置替换自旋向上电子能带图几乎相同,自旋向下电子在费米面附近由于自旋极化作用,能带图有一定变化,其中导带尤为突出,从能带图中可得出b位置替换Cr d态能带带宽较a位置替换大,说明b位置替换后,与Cr相邻的轨道间的重叠更大,成键程度更大.正是由于Cr d态能带展宽,且向低能端移动,半金属带隙较a更小.
如图6、图7,两种位置替换S原子在费米面附近几乎无变化,b位置替换由于自旋极化Cr d态Zn s态向低能端移动.正因为Cr d态向低能端移动,导致b位置替换半金属带隙更小(0.7556 eV),这同能带图分析出的结果一致.
根据能带图(如图4、图5)与态密度图(如图6、图7)可得到,a位置替换的半金属带隙更大.并且a位置替换是更稳定的基态.因此,我们将重点分析讨论a位置替换的计算结果.

表1 三元化合物ZnCrS2两不同替换位置计算结果
图4所示,三元化合物ZnCrS2在费米面附近出现自旋向上的电子态密度和自旋向下电子态密度分布劈裂,这表明体系中的电子通过交换相互作用出现了自旋有序排列.自旋向上的电子跨过了费米面,如图4(b)所示,表现出金属性;而自旋向下时,在费米面处出现一能隙(如图4(a)所示),体系还表现出半导体特性.ZnCrS2半金属带隙Eh= 0.9631 eV,这个值高于ZnxY1-xTe(x=0.25,0.75; Y=V,Cr,Mn)[25]的半金属带隙值,ZnCrS2有望成为具有更高的居里温度[25]的半金属材料.
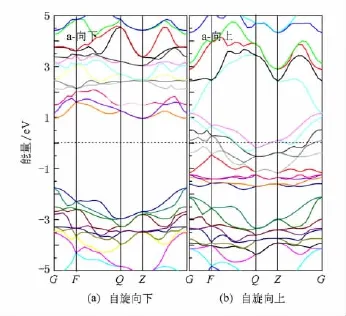
图4 ZnCrS2的能带结构(a位置替换)

图5 ZnCrS2的能带结构(b位置替换)

图6 三元化合物ZnCrS2电子态密度(a位置替换)(a)S原子分态密度;(b)Zn原子分态密度;(c)Cr原子分态密度,(d) ZnCrS2总态密度

图7三元化合物ZnCrS2电子态密度(b位置替换)(a)S原子分态密度;(b)Zn原子分态密度;(c)Cr原子分态密度;(d) ZnCrS2总态密度
图6 给出了a位置替换的ZnCrS2的态密度和分态密度.从图6中可以得到:自旋向下部分导带主要分布在0.7 eV能量范围以上,由于磁性主要表现在费米面附近,因此我们主要讨论费米面附近电子分布.在0.7—4 eV能量范围,主要来源于Cr 3 d态和少量Zn 4 s态、3p态及极少量S的3 p态、Cr 3 p态和电子的贡献;在4—5.0 eV能量范围,主要来源于Zn 3 p态的贡献,少量来自Cr 3 p态、Zn s态和S 3p态的贡献,Cr 3 d和s态亦有少量贡献.与纯Zn S相比,替换后三元化合物的态密度出现了显著的变化,出现了自旋分布劈裂,在费米面子只有一种自旋行为.在-5—-1.3 eV能量范围是上价带,其主要是S 3p电子贡献和少量Cr的3 d态贡献.在-3.4 eV处电子态密度来自于S 3p态和Cr 3d态,二者形状极其相似,由此可以判断,S原子与Cr原子存在杂化作用.
同样从图6中可以得到:自旋向上跨过费米面的电子主要由Cr 3d电子贡献,有少量S 3 p电子及极少量的Cr 3p电子贡献.-5.0—-4.8 eV能量范围,主要由S 3p和Zn 4s电子贡献,-4.8—-2.0 eV能量范围,主要由S 3 p和极少量的Cr 3 d电子贡献,Zn 4 s 3p态电子有少量的贡献,-2.0—1.5 eV能量范围,主要由Cr 3d和S 3 p电子贡献,在2.0—5.0 eV能量范围,主要由Cr 3 p电子、S 3 p,Zn 4s 3 p态电子贡献,也有少量的Cr 4 s态电子贡献.在-4.0 eV,-3.4 eV和费米面处S 3p态和Cr 3 d态存在极其相似的峰值,由此可知它们之间存在杂化作用.
为确定ZnCrS2的基态,我们分别计算了八原子体系晶胞的铁磁态和反铁磁态构型的总能量.ZnCrS2体系铁磁态的总能量为-9472.231 eV,反铁磁态的总能量为-9472.064 eV.铁磁态的总能量比反铁磁态的总能更低,计算结果表明三元化合物ZnCrS2的铁磁态是更稳定的基态.
3.3.三元化合物ZnCrS2的磁性
从表1中可得到:ZnCrS2具有磁性,模型中单个Cr原子的磁矩是4.32μB,单个S原子的磁矩是-0.16μB,单个Zn原子的磁矩为0.替换前ZnS中 Zn原子和S原子都不表现出磁性.由此可知三元化合物Zn CrS2的磁性是由于引入Cr原子后产生自旋极化状态,Cr 3d在费米面附近发生交换劈裂来实现的.
综合前面的讨论,Zn CrS2电子结构中呈现出自旋向上和自旋向下电子态密度分布,表现出半金属性;计算中得到系统总磁矩为玻尔磁矩的8倍.ZnCrS2满足半金属的两个基本特征,因此三元化合物ZnCrS2整体上呈半金属性质.且有望成为具有更高的居里温度的半金属材料.这类材料的磁性原子的密度较大,磁性强,这就使得最终器件的尺度有望较稀磁更小.
4.结论
本文采用基于密度泛函理论(DFT)的平面波超软赝势(PWPP)方法,结合广义梯度近似(GGA),计算了三元化合物ZnCrS2的能带结构、电子态密度,研究了电子结构和半金属性.通过计算发现,a位置替换的半金属能隙(0.9631 eV)较b位置替换(0.7556 eV)高,可望得到更高的居里温度;a位置替换铁磁总能量(-9472.245 eV)较b位置替换(-9472.116 eV)低,a位置替换铁磁态属更稳定的基态.三元化合物ZnCrS2能产生自旋极化状态,具有近100%的自旋极化率.能带结构和态密度及总磁矩为8.0μB都显示出半金属特征.其磁性主要是Cr原子的3 d电子在费米面附近发生交换劈裂.本文结果可望为实验研究提供指导和理论解释.
[1]Gershenfeld N A,Chuang I L 1997 Science 275 350
[2]Wolf S A,Awschalom D D,Buhrman R A,Daughton J M,von Molnár S,Roukes M L,Chtchelkanova A Y,Treger D M 2001 Science 294 1488
[3]Xing H Y,Fan G H,Zhang Y,Zhao D G 2009 Acta Phys.Sin.58 3324(in Chinese)[邢海英、范广涵、章勇、赵德刚2009物理学报58 3324]
[4]Lin Z,Guo Z Y,Bi Y J,Dong Y C 2009 Acta Phys.Sin.58 1917(in Chinese)[林竹、郭志友、毕艳军、董玉成2009物理学报58 1917]
[5]Dietl T,Ohno H,Matsukura F,Cibert J,Ferrand D 2000 Scienc 287 1019
[6]Liu Y H,Zhang L S 1994 Prog.in Phys.1 82(in Chinese)[刘宜华、张连生1994物理学进展1 82]
[7]Oiwa A,Supinski T,Munekata H 2001 Appl.Phys.Lett.78 518
[8]de Groot R A,Mueller F M 1983 Phys.Rev.Lett.50 2024
[9]Kobayashi K I,Kimura T,Sawada H,Terakura K,Tokura Y 1998 Nature 395 677
[10]Xing Y 2010 Acta Phys.Sin.59(in Chinese)[邢月2010物理学报59 in press]
[11]Lewis S P,Allen P B,Sasaki T.1997 Phys.ReV.B 55 10253
[12]Jedema F J,Filip A T,van Wees B J 2001 Nature 410 345
[13]Liu B G 2003 Phys.Rev.B 67 172411
[14]Xu Y Q,Liu B G,Pettifor D G 2002 Phys.Rev.B 66 184435
[15]Kinaga H A,Manago T,Shirai M 2000 Jpn.J.Appl.Phys.Part 2 39 L1118
[16]Mizuguchi M,Akinaga H,Manago T,Ono K,Oshima M,Shirai M,Yuri M,Lin H J,Hsieh H H,Chen C T 2002 J.Appl.Phys.91 7917
[17]Yao K L,Gao G Y,Liu Z L,Zhu L 2 0 0 5 Solid State Commun.133 301
[18]Li G N,Jin Y J,Lee J I 2010 Chin.Phys.B 19 097102
[19]Jungwirth T,Sinova J,Masek J,Kucera J,MacDonald A H 2006 Rev.Mod.Phys.78 809
[20]Shi L J,Liu B G 2007 Phys.Rev.B 76 115201
[21]Wang F,Wang X Q,Nie Z X,Cheng Z M,Liu G B 2011 Acta Phys.Sin.60(in Chinese)[王风、王新强、聂招秀、程志梅、刘高斌2011物理学报60](已接受)
[22]Fukumura T,Jin Z,Kawasaki M,Shono T,Hasegawa T,Koshihara S,Koinuma H 2001 Appl.Phys.Lett.78 958
[23]Liu Y,Liu B G 2006 J.Magn.Magn.Mater.307 245
[24]Milman V,Warren M C 2001 Journal of Physics:Condensed Matte J.Phys.-Condens.Mat.13 241
[25]Liu B G 2005 Lect.Notes Phys.676 267
PACS:63.20.dk,75.50.Cc,76.50.+g,71.20.Lp
*Project supported by the Natural Science Foundation of Chongqing(Grant No.CSTC-2007 BB4137).
Corresponding author.E-mail:xqwang@cqu.edu.cn
First-principles study on electronic structure and half-metallic ferromagnetism of ternary compound ZnCrS2*
Cheng Zhi-Mei1)2)Wang Xin-Qiang1)Wang Feng3)Lu Li-Ya1)Liu Gao-Bin1)Duan Zhuang-Fen1)Nie Zhao-Xiu1)
1)(College of Physics,Chongqing University,Chongqing 400044,China)
2)(College of Education Sciences,Chongqing Normal University,Chongqing 400047,China)
3)(College of Elementary Education,Chongqing Normal University,Chongqing 400700,China)
(Received 27 October 2010;revised manuscript received 21 November 2010)
The model of the ternary compound ZnCrS2is constructed by replacing the Zn atoms in the zinc-blend phase ZnS with Cr at two different positions a and b in a ratio of 1∶1.Then the electronic and the magnetic properties of ZnCrS2are invescigated by using the plane wave pseudopotential(PWPP)method with the density functional theory(DFT).The results show that both kinds of ZnCrS2are more stable in the ferromagnetic state than in the anti-ferromagnetic state,and are half-metallic ferromagnets with half-metallic band gaps of0.964eV and 0.755 eV,respectively.The comparison of two different models reveals that ZnCrS2in a-position replacement is more sable and also has a larger half-metallic band gap.Furthermore,the spin-polarized electronic density of states,the band structure and the magnetic moment of Zn CrS2are analyzed in detail.The present results should be useful for the future experimental study.
ZnCrS2,electronic structure,half-metallic ferromagnetism,first-principles
*重庆市自然科学基金(批准号:CSTC-2007 BB4137)资助的课题.
.E-mail:xqwang@cqu.edu.cn
