醇脱氢酶不对称还原制备手性醇的研究进展
2011-10-22王秋岩殷晓浦魏东芝
谌 容 ,王秋岩,殷晓浦,魏东芝,谢 恬
(1生物医药与健康研究中心,杭州师范大学,浙江 杭州 310012;2生物反应器工程国家重点实验室,华东理工大学鲁华生物技术研究所,上海 200237)
进展与述评
醇脱氢酶不对称还原制备手性醇的研究进展
谌 容1,2,王秋岩1,殷晓浦1,魏东芝2,谢 恬1
(1生物医药与健康研究中心,杭州师范大学,浙江 杭州 310012;2生物反应器工程国家重点实验室,华东理工大学鲁华生物技术研究所,上海 200237)
从醇脱氢酶在不对称还原制备手性醇化合物的应用出发,进行3个重要方面综述:酶分子——作为优质高效的生物催化剂,对红球菌属、乳酸菌属、赖氏菌属及嗜热微生物来源的天然或重组醇脱氢酶的催化作用进行探讨;辅酶的再生方法及优缺点分析——探索适合于特定催化反应的再生方法,以解决大规模应用时添加昂贵的辅酶带来的成本问题;体外进化技术——对目的酶活性、立体选择性与稳定性的提高。最终为酶催化制备医药及精细化工品中间体建立一条绿色高效经济的途径。
醇脱氢酶;手性中间体;辅酶再生;体外进化
近年来,随着光学异构体药物药理作用研究的深入,以及对消旋药物申报和使用的限制,手性药物的研究和开发已经成为国际新药研究的热点和方向[1]。手性是影响许多药物安全性和有效性的关键因素,研究药物及药物中间体的单一对映异构体合成越来越重要。单一对映异构体药物可以通过化学或者生物酶法合成。化学合成的难点在于昂贵的手性催化剂、对映体的分离纯化等。生物酶法合成具有高度立体异构选择性和对映体过量的优点可以弥补化学合成的缺陷,另外酶催化具有反应条件温和,副反应少、光学纯度高及环境友好等优点。因此,酶法合成手性化合物越来越受到重视[2]。
醇脱氢酶(alcohol dehydrogenase,ADH)广泛存在于各种微生物中,催化醇和醛或酮之间的相互转换,有广泛的底物特异性,同时在微生物的生理过程中起着重要的作用。根据醇脱氢酶分子长度和含金属离子性质分为3大类,typeⅠADH 的氨基酸长度在350个以上,zinc离子作为催化位点;typeⅡADH的氨基酸长度在250个左右,不需要金属离子的辅助,属于酮还原酶家族。TypeⅢ ADH需金属离子激活,在微生物的代谢中起着重要作用[3]。已报道,多种微生物来源的醇脱氢酶能将前手性酮、酮酸或酮酯还原成手性醇或手性羟基酸,而带手性羟基的化合物在药物中间体合成上有着广泛的应用[4]。来自马肝、红平红球菌、Lactobacillus brevis、Thermobacterium brockii、面包酵母的醇脱氢酶已不同程度的商业化。本文综述了近年来几种重要的醇脱氢酶在制备手性药物中间体的应用,采用醇脱氢酶催化生物转化时存在的辅酶再生的解决方案以及体外定向进化技术在醇脱氢酶性质改造上的应用。
1 微生物来源的醇脱氢酶及其催化的反应
1.1 红球菌属的醇脱氢酶
红平红球菌Rhodococcus erythropolis来源的醇脱氢酶(ReADH)是NAD(H)依赖型,属于含锌离子的中链ADH亚家族,同源四聚体,每个亚基分子量为36 kD,此酶已商业化[5]。ReADH有宽的底物特异性,对脂肪族和芳香族酮及β-酮酯都有较高的活性(表1)[6]。ReADH用于合成手性医药中间体,如合成抗HIV的蛋白酶抑制剂阿扎那韦的中间体(1S,2R)-3-氯-2-羟基-1-(苯甲基)丙基氨基甲酸,ReADH催化前手性酮还原生成(1S,2R)-醇,产量大于90%,非对映体纯度大于98%,对映体过量达到 99.4%[7]。Pollard等[8]应用此酶还原微溶于水的 3,5-双三氟甲基苯乙酮,生成(S)-3,5-双三氟甲基苯乙醇,转化率>98%和e.e.>99.9%,在最佳反应条件下,底物浓度可达到了580 mmol/L,且产物极易分离,分离率>90%,产率达到 260 g/(L·d),产物(S)-3,5-双三氟甲基苯乙醇是合成NK-1受体拮抗剂的重要手性醇中间体。ReADH能还原带庞大侧链的6-溴-β-四氢萘酮生成(S)-溴-β-四氢萘酚,能达到e.e.>99%,后者可用于合成抗心率失常候选药MK-0499[9]。ReADH还原苯基丙酮生成相应的(S)-2-羟基-苯丙醇,后者是一种胆汁固体成分分泌促进剂。(S)-2-羟基-苯丙醇也是合成苯异丙胺的中间体,是抗高血压、镇痉剂或抗癫痫药物的前体[10]。
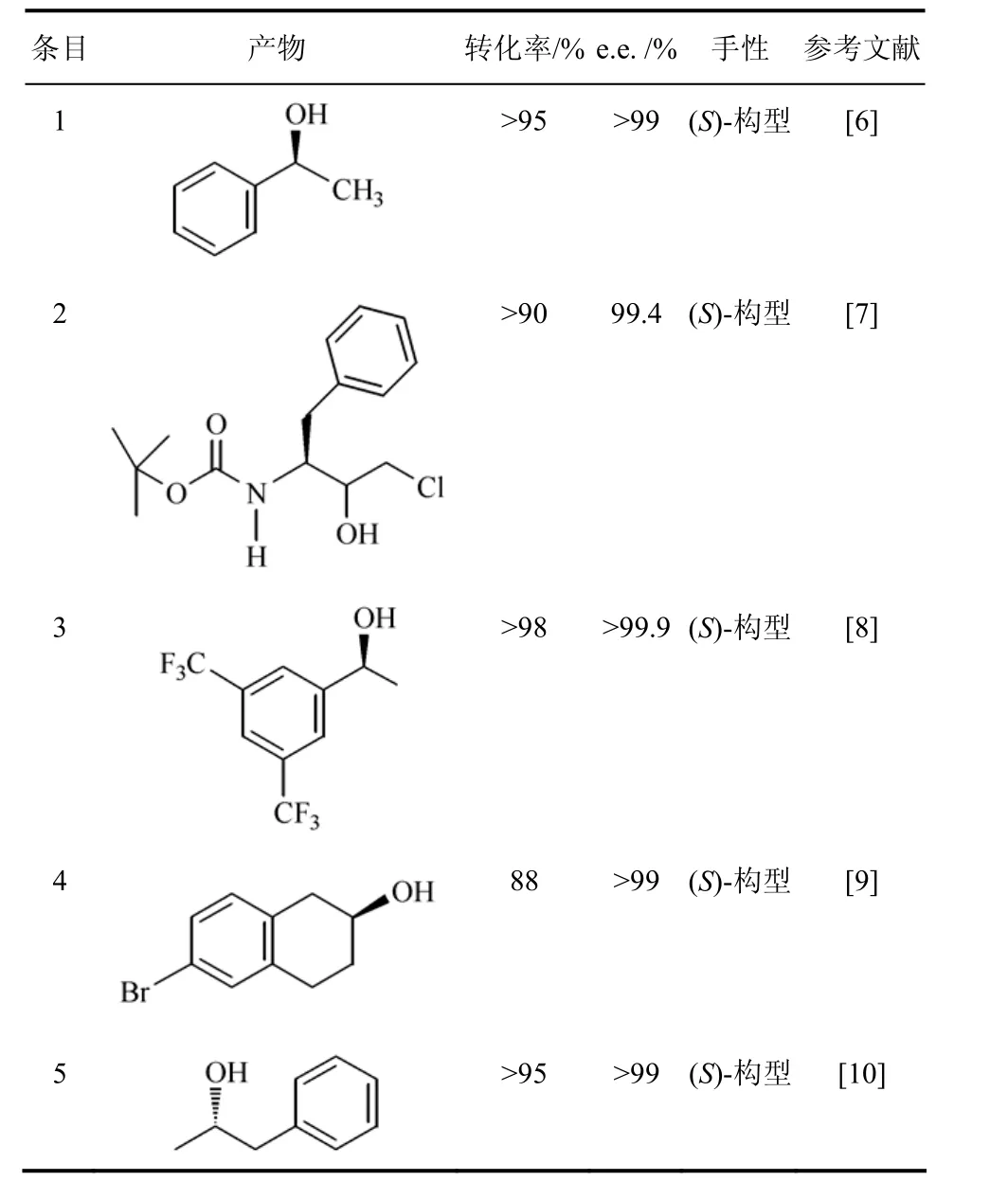
表1 Rhodococcus erythropolis的醇脱氢酶催化合成的手性醇
尽管ReADH具有宽的底物选择性及高度的立体异构性,但仍存在一些缺点,如全细胞催化时立体选择性较低,使用纯化酶作催化剂时立体选择性较好,可达e.e.>99%,且不随着反应时间和反应体系变大而降低,但异源表达量时可溶性酶量较低,多以包涵体形式存在[6]。因此,对重组 ReADH可溶性表达的进一步研究将拓宽其在不对称还原中的应用。
1.2 乳酸菌属的醇脱氢酶
研究较多的有来自Lactobacillus brevis和Lactobacillus kefir的醇脱氢酶。L. brevis醇脱氢酶(LbADH)是短链的氧化还原酶超家族的一员,同源四聚体,每个单体的分子量是26.6 kD,辅助因子是NADPH[11]。LbADH具有高度的立体专一性和对映体选择性,可以还原带有庞大侧链的酮和酮酸衍生物、β,δ-二酮基酯类和环形酮等,生成相应的(R)-醇和(R)-羟基酸酯,且e.e. >99%;具有独特的卤代苯乙酮还原活性,且活性较高[12]。Schroer等[13]建立了双膜反应工艺,LbADH还原2,5-已二酮生成(2R,5R)-己二醇,同时氧化 NAD(P)H生成NAD(P)+,反应体系中加入异丙醇实现NAD(P)H的再生,产率达到>170 g/(L·d),(2R,5R)-己二醇用于合成手性磷配体。值得一提的是LbADH能还原甲基-β-萘基甲酮生成相应的(R)-醇,且 e.e.> 99%。LbADH对甲基-β-萘基甲酮的高活性,说明该酶对侧链庞大的酮类化合物的还原能力非常强,故开展 LbADH在绿色化学合成上的应用具有重要的意义。
L. kefir的醇脱氢酶(LkADH)由759 bp核苷酸组成,编码 252个氨基酸,属于短链脱氢酶,NADPH依赖型。在大肠杆菌中重组表达的LkADH能够还原多种脂肪族、芳香族、环形的酮及β-酮酯,生成相应的(R)-醇[14-15]。Bradshaw等[16]研究大肠杆菌重组的 LkADH具有很宽的底物特异性,催化多种类型的底物还原反应生成相应的带芳香族的、环形的、多环的和脂肪族侧链的手性醇,具有较高的产率和高度的对映体过量(94%~99%)。利用L. kefir全细胞催化,细胞内的醇脱氢酶首先还原 6-氯-3,5-二氧代己酸叔丁酯的C3的酮基形成R构型的手性羟基,进一步还原C5位酮基形成S构型的手性羟基,一锅法合成6-氯-(3R,5S)-二羟基己酸叔丁酯,在22 h反应时间内产率为47.5%和e.e.>99%,(3R,5S)-二羟基己酸叔丁酯为多种HMG-CoA还原酶抑制剂的合成提供手性侧链(图1)[17]。
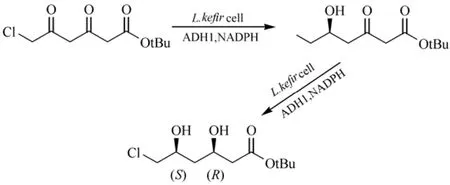
图1 L. kefir 全细胞催化一锅法合成6-氯-(3R,5S)-二羟基己酸叔丁酯
固定化细胞技术(immobilized cells)是将微生物细胞直接作为酶原,进行催化反应,具有多项优点,如节省了酶分离纯化的时间和成本,多酶促反应,增加酶的稳定性等。Tan等[18]利用固定化的L. kefir全细胞催化2,5-己二酮的还原生成(5R)-羟基己烷-2-酮,e.e.>99%,在活塞流反应器中收率达87 g/(L·d),6天后仍保持68%的残余活性。采用固定化细胞技术节约酶成本,具有应用的潜力和价值。
1.3 赖氏菌属的醇脱氢酶
赖氏菌属的Leifsoniasp.是Inoue等从苯乙烯同化后的土壤样本,对约900株菌株进行筛选所获得的能将前手性酮还原生成(S)-1-三氟苯乙醇的菌株。(S)-1-三氟苯乙醇是重要的医药中间体[19]。LeifsoniaADH的分子量是110 kD,同源四聚体,辅助因子是NADH。LeifsoniaADH可还原一系列的醛和酮,如:氯丙酮、2-庚酮、2-辛酮等前手性酮,对卤代的苯乙酮、甲氧苯乙酮、苯丁酮、环戊酮等有一定的活性,对酮酯如甲基丙酮酸、氧代丁酸盐等也有较高的活性(表2)。LeifsoniaADH催化反应具有高度的立体选择性,三氟苯乙酮还原成(S)-1-三氟苯乙醇,对映体过量大于99%,且反应体系中加入异丙醇即可实现 NADH的再生。LeifsoniaADH还原苯乙酮、双乙酰基酮、长链烷基酮等生成(R)-醇,且 e.e.>99%,有的底物转化率甚至高达100%[20-21]。带手性羟基的苯乙酮可在苯环的不同位置上进行卤代或氨基取代,产物可作为手性配体和医药中间体用于不对称合成,如用于合成拟肾上腺素药,抗生素,HIV-逆转录酶抑制剂等。
1.4 嗜热微生物的醇脱氢酶
酶的稳定性,特别是耐有机溶剂的稳定性,在工业和医药行业的应用中起着十分重要的作用。嗜热微生物来源的酶具有很高的热稳定性,而热稳定性与有机溶剂等变性剂条件下的稳定性是正相关的,因此嗜热微生物来源的醇脱氢酶吸引了众多研究者的目光[22]。
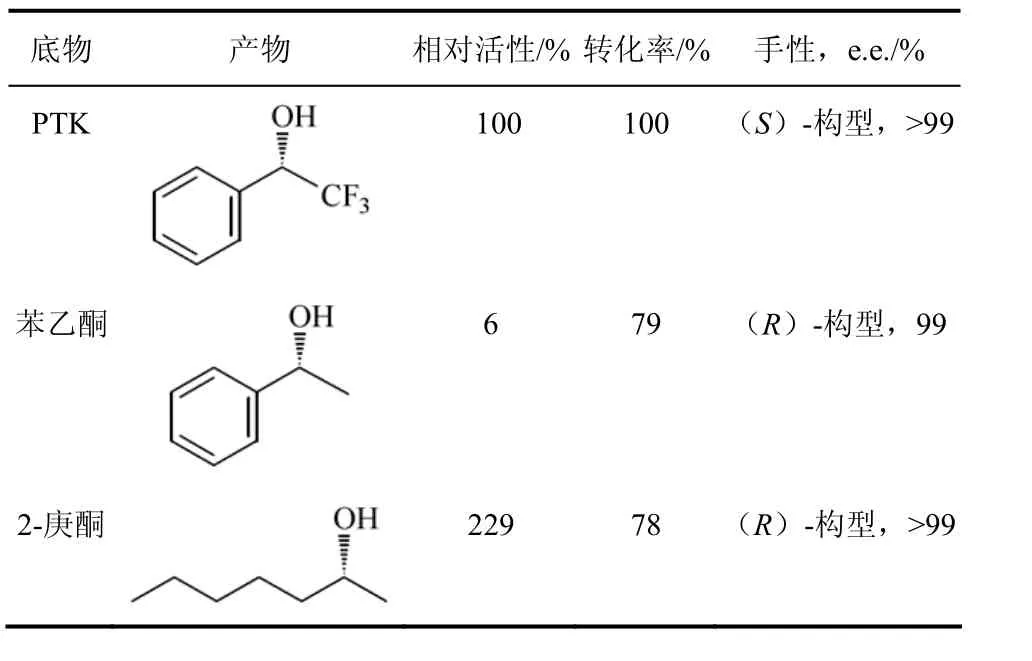
表2 Leifsonia sp. ADH的催化反应
超高温古细菌Sulfolobus solfataricus的NADH依赖的醇脱氢酶,属于中链ADH家族。其底物谱很宽,包括线性和分支的伯醇,线性和环状的仲醇和酮,并且在较高的温度(>95 ℃)仍具有活性,其高度的热稳定性在应用中具有很大的潜在价值[23]。超高温古细菌Pyrococcus furiosus的醇脱氢酶以单体存在,分子量为32 kD,在100 ℃仍有较好的活性和稳定性,对双乙酰基酮具有很好的还原能力[24]。双乙酰基酮的还原可以生成两个手性碳原子的结构,此产物可用于降血脂类他汀药物的手性侧链的合成。
嗜热微生物来源的醇脱氢酶具有较强的热稳定性和有机溶剂耐受性,在生物催化中具有较大的应用价值[25]。嗜热厌氧产乙醇杆菌Thermoanaerobacter ethanolicus的仲醇脱氢酶有很高的热稳定性,在90℃时活性最好且半衰期为1.7 h,有机溶剂稳定性非常高,如在 100%的十二烷、正辛烷、甲苯和吡啶溶液中温浴(50 ℃)3 h后活性仍分别保持90%、100%、80%和68%[26]。此酶的立体选择性与反应温度有关,如反应温度在26 ℃下对(R)-2-丁醇的氧化具有更高的催化效率;26 ℃以上时,则对(S)-2-丁醇的氧化效率较高[27]。极端嗜热菌Thermus thermophilus的短链ADH能够还原苯乙酮、2,2,2-三氟苯乙酮、四氢萘酮、α-甲基/乙基苯甲酰甲酸分别形成相应的(S)-苯乙醇(e.e.>99%)、(R)-2,2,2-三氟苯乙醇(e.e.约 93%)、(S)-四氢萘酚(e.e.>99%)、甲基(R)-扁桃酸盐(e.e.约92%)和乙基(R)-扁桃酸盐(e.e.约95%),这些化合物都是重要的医药中间体[28]。栖热菌Thermussp. ATN1的醇脱氢酶,能还原 2-戊酮、2-庚酮、苄基丙酮,苯丙酮生成(S)-2-戊醇、(S)-2-庚醇、(S)-4-苯基-2-丁醇和(S)-1-苯基-2-丙醇,且e.e.>99%。反应体系应用水-有机溶剂两相反应,均有活性,有机溶剂对酶活性影响不大,说明其耐有机溶剂稳定性很强[29]。
1.5 其它菌属的醇脱氢酶
除了上述几种研究和应用较多的醇脱氢酶,粗糙脉孢菌Neurospora crassa的ADH还原相应的底物生成(4S,6S)-5,6-二氢-4-羟基-6-甲基-4氢-噻吩并噻喃-7,7 二氧化物,后者是治疗青光眼药物trusopt 的中间体[30]。结合酵母Zygosaccharomyces rouxii的ADH可以还原3,4-亚甲二氧基苯基丙酮生成(S)-3,4-亚甲二氧基苯基丙醇,且产率>95%和e.e. >99.9%、总体上反应器的产率为75 g/(L·d),此产物是合成苯重氮基盐的重要中间体,而苯重氮基盐是重要的镇定安眠,抗焦虑药物[31]。Patel等[32-33]利用面包酵母的醇还原酶合成重要的手性药物中间体(S)-4-氯-3-羟基丁酸乙酯,后者是 HGM-CoA还原酶抑制剂和β-1,4-二氢吡啶类钙离子通道阻断剂等药物,具有重要的应用价值。面包酵母对杂环类化合物如4-乙酰吡啶、苯环及苯环衍生物等前手性酮均具有 100%的转化率和不同程度的立体异构选择性[34]。毕赤酵母的仲醇脱氢酶在大肠杆菌重组表达,能还原乙基 4-氯乙酰乙酸盐生成(S)-4-氯-3-羟基丁酸,产率为98.5%及99%对映体过量,同时表达Mycobacteriumsp. 来源的甲酸脱氢酶进行NADH的再生,实现了经济、高效、环境友好的生产方式[35]。
多种微生物来源的醇脱氢酶已经被分离,且部分醇脱氢酶的高级结构及酶学性质已经被鉴定,但醇脱氢酶是包含众多成员的超家族,其亚家族中各种类型的醇脱氢酶如长链、中链、和短链醇脱氢酶均具有不对称催化的可能(表3)。因此,若分别研究不同微生物来源醇脱氢酶的性质与功能,这无疑是一项耗时且工作量巨大的任务;且目前的研究对象为可培养微生物来源,不足环境微生物总量的1%,目前已有的研究无疑是非常小的一部分。因此,建立高效的醇脱氢酶筛选方法从整个环境微生物中快速分离到目的酶尤其重要。
2 辅酶再生系统
NAD(P) 依赖的氧化还原酶的活性依赖于NAD(P)H/ NAD(P)+,这些辅助因子价格昂贵,且稳定性受温度和pH值的影响,使其在工业使用上受到了限制,研究有效的辅助因子再生系统有重要的意义。辅酶再生是辅酶在氧化态和还原态之间的转换,可通过底物耦联法和酶耦联法来实现。辅酶再生系统的研究主要包括了 NAD(P)H的酶法再生、NAD(P)+的酶法再生、电化学再生和全细胞催化体系中辅酶的再生等[36]。辅酶再生的方法各有优劣(表4),选择的策略主要是酶在有机溶剂的稳定性、酶的活性、底物和产物对酶或细胞的影响。
目前,使用最多的是脱氢酶对NADH的再生[37],如Pfizer公司使用表皮葡萄球菌来源的(R)-乳酸-NAD氧化还原酶与来自Candida boidinii的甲酸脱氢酶联合酶催化在工业级规模级别上实现了(R)-3-(4-氟苯基)-2-羟基丙酸的生产[38]。经济、高活性且稳定的葡萄糖脱氢酶或6-磷酸葡萄糖脱氢酶催化的NAD(P)H再生系统已有报道[39]。亚磷酸脱氢酶也可用于NAD(P)的再生,但其高磷废物的产生和反应体系依赖pH值的劣势限制了其应用[40]。Lopez de Felipe等[41]在L. lactis中表达了NADH氧化酶,将NADH脱氢氧化生成NAD+,从而实现了NAD+的再生。电化学法不需要酶和底物,因此不会有产物和副产物,具有清洁、可持续的优点,被认为是有力的绿色技术。如铑复合物与Thermussp.来源的醇脱氢酶在有机/水两相反应体系中实现NADH的再生,获得(1S,3S)-3-甲基环戊醇,为电化学法在有机合成应用中提供了有力的证据[42];锡氧化物电极在多种反应条件下都可用于 NADH的氧化和NAD+的还原。Kim等[43]使用醇脱氢酶氧化异丙醇生成丙酮,同时还原NAD+生成NADH,锡氧化物电极再将NADH氧化成NAD+,从而实现
了NAD+的循环,此电化学方法对NADP+的循环再生同样具有高效的作用。光化学法依赖光合细菌实现辅酶因子的再生循环,但效率低[44]。醇脱氢酶不仅可以催化底物向目标产物的转化,同时也可用于辅助因子的还原,从而实现单酶在同一个反应体系中起两种作用[45]。氢化酶可还原H2,同时实现NAD(P)的循环再生,但H2的易燃性是整个反应存在不安全的因素[46]。因此,选择合适的辅酶再生系统是醇脱氢酶催化反应的重要因素,通过再生循环的建立,可实现辅助因子经济有效的再生。

表3 上述几种微生物来源的醇脱氢酶性质特性

表4 辅酶再生方法
3 酶的体外进化
酶作为生物催化剂其专一性和高效性是一般催化剂所不能比拟的,且具有环境友好性的优势。但是天然酶的许多性质,如较差的选择性、不稳定性以及苛刻的酶促反应条件,使其不适应工业大规模应用,因此对酶进行定向进化研究成为根据实际需要改变酶的结构和性质的有效途径[47-48]。
对醇脱氢酶的体外进化,主要包括提高酶对底物的活力、增加底物特异性、提高有机溶剂稳定性、降低对辅基的作用和改变辅酶亲和力。通过易错PCR构建超高温古生菌Pyrococcus furiosus醇脱氢酶的突变库,获得在常温条件下还原2,5-己二酮生成(2S,5S)-己二醇的突变体,其活力提高至野生菌的 10倍[49]。通过理性设计嗜热厌氧产乙醇杆菌的仲醇脱氢酶,获得单氨基酸突变的菌株(I86A),此突变株的活性位点比野生型能容纳更大的空间位阻取代基,而且产物的立体化学选择是按照anti-Prelog的构型模式进行的[50]。W110A 突变株能还原相应的酮生成(S)-1-苯基-2-丙醇、(S)-4-苯基-2-丁醇,且e.e.>99%,而其野生型无活性[51]。对来源于Rhodococcussp. ST-10的苯乙醛还原酶(一种中链的醇脱氢酶)进行易错PCR建立随机突变文库,筛选获得了以异丙醇作为底物进行NADH的再生的突变株,建立了单酶催化两个反应,同时实现底物向产物的转化以及辅酶的再生循环[45]。某些醇脱氢酶反应体系中需要一些金属离子作用,基于易错PCR的随机突变文库筛选不受Mg2+等离子限制的醇脱氢酶有利于更好的应用[52]。L. brevis的醇脱氢酶依赖NADP(H),对 NAD(H)的依赖较小。但NADP(H)比 NAD(H)昂贵且不稳定,因此Machielsen 等对其进行计算机模拟设计以构建突变文库,获得相对野生菌活性高4倍的突变菌株(对NADH的亲和力),此突变菌株(R38P)已证明具有更有用的药物前体合成作用[53]。酶的定向进化是用于解决酶工程中重大问题的关键技术,使酶的催化性能达到和接近工业用途的要求逐渐成为可能。
尽管科研工作者在使用易错 PCR技术或定点突变技术获得了酶学性质改进的突变株,但是此技术工作量大及消耗时间长,且得到的突变株活性提高程度有限。本课题组成员利用传统的活性筛选方法从宏基因组文库中筛选到新酶,并以此为模板设计引物采用改进的PCR技术钓取大量的同源序列,结合 gene shuffling获得了具有活性的多条基因序列,为根据需要而快速、有效地筛选或改进酶学性质发挥了重要的作用,该成果发表在Journal of Biological Chemistry杂志[54]。
4 结语与展望
在工业级生物催化应用中占主导地位的是氧化还原酶(30%),仅次于水解酶(44%)。醇脱氢酶正是重要的一种氧化还原酶。尽管醇脱氢酶在医药、精细化工和环境保护等行业中的巨大作用,但其实际应用绝大多数仍处在实验室阶段,要尽快实现其工业化应用,需要解决以下问题。
(1)尽管有多种微生物来源的酶被分离并鉴定,但是可培养微生物不足微生物总数的 1%。因此开展各种生境环境中未培养微生物来源的酶的分离与鉴定,无疑会为生物催化的重要元素即酶提供大量的来源,从而有更高的概率筛选到更适合工业化应用的“理想”酶。本课题组成员从宏基因组出发,采用基于序列筛选的方法获得新酶,使酶的来源突破了传统的可培养微生物的局限[55]。
(2)利用体外重组制备醇脱氢酶成本高,而全细胞催化又受制于底物利用率及细胞毒性的问题。
(3)由于大多数底物是有机试剂,微溶于水或不溶于水,导致反应体系中底物利用率低,且酶的稳定性降低。
(4)辅助因子的添加从而增加反应的成本[56]。
针对上述问题,获得高稳定性的酶并解决反应体系中辅酶再生循环这两方面显得尤其重要。同时,从重组酶和基因工程菌两方面着手解决醇脱氢酶在工业化应用中的难点,将会大大加快其应用。更重要的是,近几年发展的非水介质中的酶催化反应和酶及细胞的固定化技术将会提高酶的工业化应用[57],为实现其合成医药中间体的大规模转化奠定基础。
[1]FDA’s policy statement for the development of new stereoisomeric drugs [J].Chirality,1992,4(5):338-340.
[2]Goldberg K,Schroer K,Lütz S,et al. Biocatalytic ketone reduction—a powerful tool for the production of chiral alcohols—partⅠ:Processes with isolated enzymes[J].Appl. Microbiol. Biotechnol.,2007,76(2):249-255.
[3]Radianingtyas H,Wright P C. Alcohol dehydrogenases from thermophilic and hyperthermophilic archaea and bacteria[J].FEMS Microbiol. Rev.,2003,27(5):593-616.
[4]Patel R N. Biocatalytic synthesis of chiral intermediates[J].Food Technol. Biotechnol.,2004,42(4):305-325.
[5]Abokitse K,Hummel W. Cloning,sequence analysis,and heterologous expression of the gene encoding a(S)-specific alcohol dehydrogenase fromRhodococcus erythropolisDSM 43297[J].Appl. Microbiol. Biotechnol.,2003,62:380-386.
[6]Hummel W,AbokitseK,Drauz K,et al. Towards a large-scale asymmetric reduction process with isolated enzymes:Expression of an(S)-alcohol dehydrogenase inE. coliand studies on the synthetic potential of this biocatalyst[J].Adv. Synth. Catal.,2003,345:153-159.
[7]Patel R,Chu L,Mueller R. Diastereoselective microbial reduction of(S)-[3-chloro-2-oxo-1-(phenylmethyl)propyl]carbamic acid,1,1-dimethylethyl ester[J].Tetrahedron:Asymmetry,2003,14(20):3105-3109.
[8]Pollard D,Truppo M,Pollard J,et al.Effective synthesis of(S)-3,5-bistrifluoromethylphenyl ethanol by asymmetric enzymatic reduction[J].Tetrahedron:Asymmetry,2006,17:554-559.
[9]Hussain W,Pollard D J,Truppo M,et al. Enzymatic ketone reductions withco-factor recycling:Improved reactions with ionic liquidco-solvents[J].J. Mol. Catal. B:Enzym.,2008,55:19-29.
[10]Liese A,Seelbach K,Wandry C. Industrial Biotransformations[M]. New York:Wiley-VCH,2000:423.
[11]Niefind K,Müller J,Riebel B,et al. The crystal structure of R-specific alcohol dehydrogenase fromLactobacillus brevissuggests the structural basis of metal dependency[J].J. Mol. Biol.,2003,327:317-328.
[12]Wolberg M,Hummel W,Müller M. Biocatalytic reduction of b,ddiketo esters:A highly stereoselective approach to all four stereoisomers of a chlorinated b,d-dihydroxy hexanoate[J].Chem. Eur. J.,2001,7:4562-4571.
[13]Schroer K,Lutz S. A continuously operated bimembrane reactor process for the biocatalytic production of(2R,5R)-hexanediol[J].Org. Process Res. Dev.,2009,13:1202-1205.
[14]Weckbecker A,Hummel W. Cloning,expression,and characterization of an(R)-specific alcohol dehydrogenase fromLactobacillus kefir[J].Biocatal. Biotransform.,2006,24(5):380-389.
[15]Humme1 W. Reduction of acetophenone to R(+)-phenylethanol by a new alcohol dehydrogenase fromLactobacillus kefir[J].Appl. Microbio. Biotechnol.,1990,34(1):15-19.
[16]Bradshaw C W,Humme1 W,Wong C H.Lactobacillus kefiralcohol dehydrogenase:A useful catalyst for synthesis[J].J. Org. Chem.,1992,57(5):1552-1556.
[17]Pfruender H,Amidjojo M,Hang F,et al. Production ofLactobacillus kefircells for asymmetric synthesis of a 3,5-dihydroxycarboxylate[J].Appl. Microbiol. Biotechnol.,2005,67:619-622.
[18]Tan AW I,Fischbach M,Huebner H,et al.Synthesis of enantiopure(5R)-hydroxyhexane-2-one with immobilised whole cells ofLactobacillus kefiri[J].Appl. Microbiol. Biotechnol.,2006,71:289-293.
[19]Inoue K,Makino Y,Itoh N. Purification and characterization of a novel alcohol dehydrogenase fromleifsoniasp. strain s749:A promising biocatalyst for an asymmetric hydrogen transfer bioreduction[J].Appl. Environ. Microbiol.,2005,71(7):3633-3641.
[20]Inoue K,Makino Y,Dairi T,et al. Gene cloning and expression of leifsonia alcohol dehydrogenase(lsadh)involved in asymmetric hydrogen-transfer bioreduction to produce(R)-form chiral alcohols[J].Biosci. Biotechnol. Biochem.,2006,70(2):418-426.
[21]Inoue K,Makino Y,Itoh N. Production of(R)-chiral alcohols by a hydrogen-transfer bioreduction with NADH-dependentLeifsoniaalcohol dehydrogenase(LSADH)[J].Tetrahedron:Asymmetry,2005,16:2539-2549.
[22]Burton S G,Cowan D A,Woodley J M. The search for the ideal biocatalyst[J].Nat. Biotechnol.,2002,20:37-45.
[23]Rella R,Raia C A,Pensa M. A novel archaebacterial NAD+-dependent alcohol dehydrogenase[J].Purification and Properties. Eur. J. Biochem.,1987,167:475-479.
[24]Machielsen R,Uria A R,Servé W M,et al. Production and characterization of a thermostable alcohol dehydrogenase that belongs to the aldo-keto reductase superfamily[J].Appl. Environ. Microbiol.,2006,72(1):233-238.
[25]Wolberg M,Filho M V,Bode S. Chemoenzymatic synthesis of the chiral side-chain of statins:Application of an alcohol dehydrogenase catalysed ketone reduction on a large scale[J].Bioprocess Biosyst. Eng.,2008,31(3):183-191.
[26]Miroliaei M,Nemat-Gorgani M. Effect of organic solvents on stability and activity of two related alcohol dehydrogenases:A comparative study[J].Int. J. Biochem. Cell Biol.,2002,34:169-175.
[27]Pham Van T,Phillips R S. Effects of substrate structure and temperature on the stereospecificity of secondary alcohol dehydrogenase from thermoanaerobacter ethanolicus[J].J. Am. Chem. Soc.,1990,112:3629-3632.
[28]Pennacchio A,Pucci B,Secundo F. Purification and characterization of a novel recombinant highly enantioselective short-chain NAD(H)-dependent alcohol dehydrogenase fromThermus thermophilus[J].Appl. Environ. Microbiol.,2008,74(13):3949-3958.
[29]Hollrigl V,Hollmann F,Kleeb A C. TADH,the thermostable alcohol dehydrogenase fromThermussp. ATN1:A versatile new biocatalyst for organic synthesis[J].Appl. Microbiol. Biotechnol.,2008,81(2):263-273.
[30]Blacklock T J,Sohar P,Butcher J W,et al. An enantioselective synthesis of the topically-active carbonic anhydrase inhibitor MK-0507:5,6-dihydro-(S)-4-(ethylamino)-(S)-6-mehtyl-4H-thieno[2,3-beta]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride[J].J. Org. Chem.,1993,58:1672-1679.
[31]Vicenzi J T,Zmijewski M J,Reinhard M R,et al.Large-scale stereoselective enzymatic ketone reduction within-situproduct removalviapolymeric adsorbent resins[J].Enzyme Microb. Technol.,1997,20:494-499.
[32]Yasohara Y,Kizaki N,Hasegawa J. Synthesis of optically active ethyl 4-chloro-3-hydroxybutanoate by microbial reduction[J].Appl. Microbiol. Biotechnol.,1999,51(6):847-851.
[33]Lüdeke S,Richter M,Müller M. Stereoselective synthesis of three iso-mers of tert-butyl 5-hydroxy-4-methyl-3-oxohexanoate through alcohol dehydrogenase-catalyzed dynamic kinetic resolution[J].Adv. Synth. Catal.,2009,351:253-259.
[34]Bawa R A,Ajjabou F,Shalfooh E. Enzymatic reduction of ketones to optically active secondary alcohols[J].Journal of Physical Science,2008,19(2):1-5.
[35]Matsuyama A,Yamamoto H,Kobayashi Y. Practical application of recombinant whole-cell biocatalysts for the manufacturing of pharmaceutical intermediates such as chiral alcohols[J].Org. Proc. Res. Dev.,2002,6(4):558-561.
[36]van der Donk W A,Zhao H. Recent developments in pyridine nucleotide regeneration[J].Curr. Opin. Biotechnol.,2003,14:421-426.
[37]Tishkov V I,Popov V O . Protein engineering of formate dehydrogenase [J].Biomol. Eng.,2006,23:89-110.
[38]Tao J H,Org McGee K. Development of a continuous enzymatic process for the preparation of(R)-3-(4-fluorophenyl)-2-hydroxy propionic acid[J].Process Res. Dev.,2002,6:520-524.
[39]Weckbecker A,Hummel W. Glucose dehydrogenase for the regeneration of NADPH and NADH[M]. In:Barredo JL(ed)Microbial Enzymes and Biotransformations. Humana,Totowa,NJ,2005:225-238.
[40]Johannes T W,Woodyer R D,Zhao H. Efficient regeneration of NADPH using an engineered phosphite dehydrogenase[J].Biotechnol. Bioeng.,2007,96(1):18-26.
[41]Lopez de Felipe F,Kleerebezem M,de Vos W M,et al. cofactor engineering:A novel approach to metabolic engineering inLactococcus lactisby controlled expression of NADH oxidase[J].J. Bacteriol.,1998,180(15):3804-3808.
[42]lrigl V H,Otto K,Schmid A. Electroenzymatic asymmetric reduction of rac-3-methylcyclo-hexanone to(1S,3S)-3-methylcyclohexanol in organic/aqueous media catalyzed by a thermophilic alcohol dehydrogenase[J].Adv. Synth. Catal.,2007,349,1337-1340.
[43]Kim Y H ,Yoo Y J. Regeneration of the nicotinamide cofactor using a mediator-free electrochemical method with a tin oxide electrode[J].Enzyme Microb. Technol.,2009,44:129-134.
[44]Willner I,Mandler D. Enzyme-catalysed biotransformations through photochemical regeneration of nicotinamide cofactors[J].Enzyme Microb. Technol.,1989,11(8):467-483.
[45]Makino Y,Dairi T,Itoh N. Engineering the phenylacetaldehyde reductase mutant for improved substrate conversion in the presence of concentrated 2-propanol[J].Appl. Microbiol. Biotechnol.,2007,77:833-843.
[46]Kroutil W,Mang H,Edegger K,et al. Recent advances in the biocatalytic reduction of ketones and oxidation of sec-alcohols[J].Curr. Opin. Chem. Biol.,2004,8(2):120-126 .
[47]蒋本国,范圣第,刘宝全. 酶的定向进化[J]. 化学通报,2004,67(5):394.
[48]Böttcher D,Bornscheuer U T. Protein engineering of microbial enzymes[J].Curr. Opin. Microbio.,2010,13:1-9.
[49]Machielsen R,Leferink N G,Hendriks A,et al. Laboratory evolution ofPyrococcus furiosusalcohol dehydrogenase to improve the production of(2S,5S)-hexanediol at moderate temperatures[J]. Extremophiles,2008,12(4):587-594.
[50]Musa M,Lott N,Laivenieks M,et al. A single point mutation reverses the enantiopreference of thermoanaerobacter ethanolicus secondary alcohol dehydrogenase[J].Chem. Cat. Chem.,2009,1:89-93.
[51]Ziegelmann-Fjeld K I,Musa M M,Phillips R S,et al. Thermoanaerobacter ethanolicus secondary alcohol dehydrogenase mutant derivative highly active and stereoselective on phenylacetone and benzylacetone[J].Protein Engineering,Design & Selection,2007,20(2):47-55.
[52]Ho K K,Hurley T D,Weiner H. Selective alteration of the rate-limiting step in cytosolic aldehyde dehydrogenase through random mutagenesis[J].Biochemistry,2006,45(31):9445-9453.
[53]Machielsen R,Looger L L,Raedts J,et al. Cofactor engineering ofLactobacillus brevisalcohol dehydrogenase by computational design[J].Eng. Life Sci.,2009,9(1):38-44.
[54]Wang Q,Wu H,Wang A,et al. Prospecting metagenomic enzyme subfamily genes for dna family shuffling by a novel pcr-based approach[J].J. Biol. Chem.,2010,285(53):41509-41516.
[55]谌容,王秋岩,杨兵,等. 温泉环境宏基因组文库中醛脱氢酶基因的克隆及鉴定[J]. 杭州师范大学学报:自然科学版,2010,8(4):286-289.
[56]王普,祖蕾,何军邀,等. 基因工程菌在不对称还原制备手性醇中的应用进展[J]. 化工进展,2008,27(7):977-1002.
[57]刘庆彬. 酶催化工艺用于制药工业的研究进展[J]. 化工进展,2004,23(6):590-594.
Progress of asymmetric reduction with alcohol dehydrogenase for preparation of chiral alcohols
CHEN Rong1,2,WANG Qiuyan1,YIN Xiaopu1,WEI Dongzhi2,XIE Tian1
(1Center for Biomedicine and Health,Hangzhou Normal University,Hangzhou 310012,Zhejiang,China;2State Key Laboratory of Bioreactor Engineering,New World Institute of Biotechnology,East China University of Science and Technology,Shanghai 200237,China)
For preparative applications in asymmetric reduction with alcohol dehydrogenase,three issues have to be considered:An appropriate enzyme,an efficient coenzyme-regenerating method and a suitable in vitro evolution technique. Enzyme is the source of effective biocatalyst. The native or recombinational alcohol dehydrogenases fromRhodococcussp.,Lactobacillussp.,Leifsoniasp. and thermophilic microbiology,and their catalysis were analyzed. The advantages and disadvantages of coenzyme-regeneration methods were discussed. The efficient coenzyme-regeneration can decrease costs for large-scale application. In vitro evolution technique can improve the activity,stability and selectivity of target enzyme. The final aim is to establish effective and green ways to obtain chiral drug intermediates by enzymatic synthesis.
alcohol dehydrogenase;chiral Intermediates;coenzyme regeneration;in vitro evolution
Q 599
A
1000–6613(2011)07–1562–08
2011-01-10;修改稿日期:2011-02-24。
国家自然科学基金(21006018)及浙江省科技厅(2009C31086)及杭州师范大学中青年培育基金(2010QN19)项目。
谌容(1984—),女,博士研究生。E-mail rchen1984@ 163.com。联系人:谢恬,教授,硕士生导师,E-mail tianxie.hznu@ gmail.com。
