丙戊酸钠对APP/PS1双重转基因AD模型小鼠脑内老年斑和神经元的影响☆
2011-09-14龙志敏赵蕾高宝兵汪克建贺桂琼
龙志敏 赵蕾 高宝兵 汪克建 贺桂琼
阿尔茨海默病(Alzheimer’s disease,AD)是一 种进行性的不可逆的神经退行性疾病,现已成为严重威胁老年人健康的四大疾病之一,是临床最为常见的痴呆类型。神经细胞外β淀粉样蛋白(amyloid β peptide,Aβ)沉积形成的老年斑(senile plaques,SP)、细胞内tau蛋白过度磷酸化聚集形成的神经原纤维缠结(neurofibrillary tangles,NFT)、皮质和海马的神经元丢失是AD脑的三大典型病理特征。然而,关于AD的病理过程,目前尚无一个学说能全面解释,这导致AD的治疗仍缺乏特异性。近期一系列研究发现,糖原合成酶激酶-3(glycogen synthase kinase 3,GSK-3) 同时参与 AD病理特征SP和NFT的形成及神经元凋亡[1]。作为GSK-3β的选择性抑制剂,丙戊酸钠(valproic acid sodium salt,VPA)是否对AD患者脑组织起神经保护作用,本课题拟用VPA处理APP/PS双重转基因AD模型小鼠,结合免疫组化、甲硫素S染色、Nissl染色、TUNEL染色、ELISA定量检测来探讨VPA的神经保护作用及相关机制。
1 材料与方法
1.1 研究对象 APP/PS1双重转基因传代小鼠的建立:将购于Jackson Lab雄性和雌性的APP/PS1双转基因杂合体种鼠B6C3-Tg(APPswe,PSEN1de9)85 Dbo/J合笼,交配繁殖。待产下的后代达3周龄时用PCR进行基因分型。
1.2 实验方法
1.2.1 基因分型 APP/PS1双重转基因小鼠的鉴定:剪子代小鼠鼠尾约0.5 cm,按照DNA提取试剂盒(Tiangen公司)步骤提取基因组DNA,核酸蛋白测定仪测定浓度后进行PCR扩增,1.5%Agrose gel电泳检测,紫外分析仪下观察同时出现APP,PS1条带的即为APP/PS1双转基因小鼠(见图1)。
PCR扩增:根据提供的引物扩增APP和PS1(以β-actin为内对照)。APP引物上游 5′-CACCA CAGAATCCAAGTCGG-3′,下游 5′-CTTGACGTTCT GGCCTCTTCC-3′;PS1 引物上游 5′-CAGGTGCTAT AAGGTCAT-3′,下游 5′-ATCACAGCCAAGATGAGC-3′;β-actin 引物上游 5′-GACAGGATGCAGAAGGA GAT-3′,下游 5′-TTGCTGATCCACATCTGCTG-3′。反应条件:94℃ ×5 min→(94℃ 30 s→ 58℃ 40 s→ 72℃ 40 s) 共 35个循环→72℃ 10 min(-20℃保存PCR产物)。APP基因PCR产物长350 bp,PS1 扩增片段长 608 bp,β-actin 为 446 bp[2]。
1.2.2 动物分组及药物处理 待APP/PS1鉴定为阳性的小鼠取4月龄APP/PS1双重转基因小鼠24只,随机分为A、B两组(每组12只)。A组小鼠给予 VPA 腹腔注射,VPA 用药量:30 mg/(kg·d),用无菌生理盐水稀释(4℃保存),腹腔注射,持续4周;B组注射等量生理盐水作对照,每天上午固定时间给药。所有实验遵循重庆医科大学实验动物使用相关伦理要求。
1.2.3 动物标本的制备 小鼠在药物注射结束后,经2%水合氯醛麻醉,断头,冰上取脑,左侧脑组织放入4%多聚甲醛固定72 h,30%蔗糖脱水,待组织块沉底后,恒冷切片机冠状位连续切片,片厚 20 μm,切片入 1×PBST中,一部分切片采用漂浮法作免疫组化和Thioflavin S染色,一部分裱片留作Nissl染色和TUNEL染色。右侧脑组织加蛋白裂解液匀浆,于4℃ 低温以12000 r/min离心10 min,取上清保存于-80℃冰箱,用于 ELISA测定。
1.2.4 免疫组化染色 切片入88%甲酸,室温下溶解老年斑中的不溶性Aβ 20 min,1×PBST漂洗5 min×3次,5%正常山羊血清室温封闭60 min,转移至一抗 4G8(浓度 1∶500),4℃48~72h,1×PBST漂洗 5 min× 3 次,加入 ABC 混合液(1∶1000),室温30 min,1×PBST漂洗5 min×3次,DAB显色后裱片,梯度酒精脱水,二甲苯透明,封片后光镜下观察。
1.2.5 Thioflavin S染色 切片浸入丙酮,室温10 min,依次用70%和80%酒精、双蒸水各冲洗30 s,0.1%KMNO4染色30 s,双蒸水、70%和80%酒精各冲洗 30 s,0.1%Thioflavin S(溶于 80%乙醇)室温染色15 min,80%和70%酒精、双蒸水各冲洗30 s,裱片,水溶性封片剂封片后荧光显微镜下观察。
1.2.6 原位末端标记(TUNEL)检测 按照凋亡试剂盒(Roche公司)说明进行操作:新制备的4%多聚甲醛室温固定切片30 min,0.3%H2O2甲醇溶液室温孵育30 min,0.1%TritonX-100在冰浴中孵育2 min,滴加TUNEL反应混合物,37℃,60 min,滴加转化剂-POD,37℃,30 min,每一步骤后均PBS洗片5 min×3次,DAB显色后光镜下观察。结果判断:棕黄色为阳性反应颜色。每张切片选5个高倍视野计数阳性表达数。
1.2.7 ELISA 定量测定 Aβ40、Aβ42 的含量 按照ELISA试剂盒(R&D公司)说明操作:将试剂盒及待测标本置于室温20~30 min,配制标准品浓度为:32、16、8、4、2 pg/mL,待作标准曲线。 取出已包被的96孔板,在标准品孔中加入稀释好的标准品50 μL,在待测样品孔中加样品稀释液 40 μL和脑组织裂解样品10 μL,半透膜封存,37℃温育30 min。洗板,加酶标试剂 50 μL,37℃温育 30 min。洗板,加显色剂 A、B 各 50 μL,37℃避光显色 10 min,终止反应,450 nm波长下读取吸光度值,根据标准曲线计算Aβ40和Aβ42浓度。
1.3 统计学方法 采用SPSS 17.0统计软件对实验数据进行独立样本t检验分析,实验中形态学染色显示的两组小鼠脑内SP的数量、凋亡神经元数量及ELISA定量测得的Aβ水平均以x±s表示,检验水准 α = 0.05。
2 结果
2.1 APP/PS1双转基因小鼠的鉴定 雌性和雄性的种鼠产下后代,剪鼠尾提取基因组DNA经PCR扩增后,1.5%琼脂糖电泳结果同时出现APP和PS1的条带即为APP/PS1双转基因阳性小鼠(图 1)。
2.2 VPA处理后小鼠脑内老年斑的变化
2.2.1 免疫组织化学结果显示 VPA处理组和生理盐水对照组小鼠大脑皮质及海马等区域均出现4G8阳性斑块(即SP),说明两组小鼠脑组织都有Aβ沉积。对照组小鼠脑内4G8阳性斑块数量多,体积大;VPA处理组斑块数量明显减少,体积明显缩小(图2A)。取每只小鼠不同部位的5张脑片进行阳性斑块计数,结果显示:A组小鼠大脑皮质及海马阳性斑块数(单位:个)为(15.67 ± 2.52),明显少于 B 组对照组(42 ± 5.29)(t= 7.78,P < 0.01),差异具有显著统计学意义。

图1 APP/PS1双转基因小鼠的基因分型
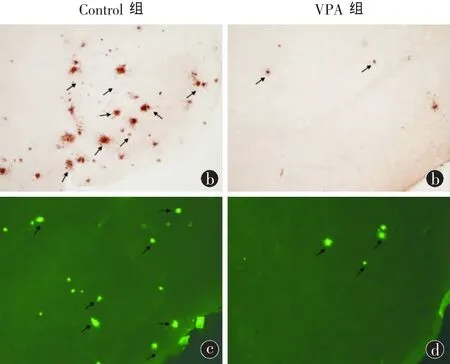
图2 两组小鼠大脑皮质老年斑(SP)比较。a、b 4G8免疫组化染色 ×100,c、d Thioflavin S染色 100×
2.2.2 Thioflavin S染色结果显示,生理盐水对照组小鼠大脑皮质及海马等区域出现大量绿色荧光斑块,而VPA处理组斑块明显减少,与免疫组织化学的结果一致(图2B)。
2.3 VPA对神经元的影响
2.3.1 Nissl染色结果 脑组织切片呈蓝紫色,两组均出现大量肿胀变圆球状细胞,突触消失,胞浆中可见尼氏小体明显减少,出现较多凋亡或凋亡前期细胞,但VPA组中存在的正常神经元多于对照组,且VPA处理组在皮质区尤其是内嗅区单位面积神经元数目较生理盐水对照组明显增多,海马CA各区厚度比对照组小鼠增厚,排列更整齐致密(图 3)。
2.3.2 TUNEL染色结果 显示两组小鼠脑内神经细胞均可见明显染色阳性的凋亡细胞,细胞轮廓清楚,细胞核深染呈棕黄色,由于细胞凋亡时细胞核的DNA可渗透到胞浆,所以部分细胞胞浆也着色。VPA 治疗组小鼠皮质凋亡细胞(144.33±24.13)明显少于对照组(81 ± 24.43)(t= 5.95,P < 0.05),有显著统计学意义(图4)。
2.4 ELISA定量检测脑内Aβ水平
ELISA 结果显示,VPA 治疗组 Aβ40(777.50 ±161.58)pg/mL、Aβ42(232.50 ± 117.76)pg/mL 的生成较对照组 [Aβ40:(1092.5 ± 84.90)pg/mL,Aβ42:(925.00 ± 219.09)pg/mL]显著减少(t分别为 4.23和 7.51,P < 0.05),且 Aβ42 减少更加明显,差异具有统计学意义。

图3 Nissl染色示皮质海马区神经元的比较。皮质出现较多凋亡或凋亡前体细胞,但VPA组(b)的正常神经元多于对照组(a),且 VPA 组的皮质区(d)、海马区(f)的神经元密度大于对照组相应脑区(c,e)。(a、b、c、d 400 ×,e、f 100 ×)

图4 TUNEL染色示两组小鼠皮质凋亡细胞的比较(400×),n=9
3 讨论
GSK-3是一种多功能的蛋白激酶。研究表明,GSK-3除参与糖代谢外,还参与细胞内其他重要生理过程,如细胞的分化、增殖和凋亡及信号转导等[3]。 GSK-3 主要有 2 种亚型:GSK-3a 和 GSK-3β。其中GSK-3β活性调节异常与人类多种疾病有关,如糖尿病、AD 等[4]。 近期一系列研究发现,GSK-3β与AD病理特征SP和NFT的形成及神经元凋亡密切相关。因此学者提出了AD发病机制的新学说,即“GSK-3β 学说”[5],认为 GSK-3β 在 AD 发病机制中起核心作用。可见,GSK-3β是AD的一个潜在性治疗靶点,开展以GSK-3β为靶点的药物研究将为AD的有效防治开辟新途径。
VPA是一种在临床上得到广泛应用的广谱抗癫痫药物,近年对VPA的研究已突破了抗癫痫的范畴,研究结果发现VPA还具有神经保护和抗凋亡作用。最近的一项研究发现[6],VPA是一种选择性的GSK-3β抑制剂。因此,运用VPA治疗AD便具有了可能性。
研究表明,人类APP和PS1基因突变与AD发病相关,转入人类APP/PS1突变基因小鼠表现出与AD患者相似的病理与记忆缺陷,是一种理想而可靠的AD转基因动物模型。与APP单转基因小鼠相比,APP/PS1双转基因小鼠的优点是发病更早、病变率更高、老年斑更明显,因此该转基因小鼠已被应用于AD病因、发病机制及防治药物的研究和开发[7]。
在AD的发病机制中,Aβ沉积是其中心环节。Aβ是由其前体蛋白APP分别经β-、γ-分泌酶相继作用产生。脑内Aβ的主要形式是Aβ40和Aβ42,正常情况下二者的生成比例为 10∶1。 其中Aβ42极易纤维化聚集,形成具有神经毒性的低聚体。 当脑内 Aβ40/Aβ42 比例失调、Aβ42 产生过量,便导致具有神经毒性的Aβ42在脑内聚集、沉积,最终形成老年斑。老年斑主要分布在大脑皮质及海马等与学习记忆密切相关的脑区。大量研究表明,Aβ(尤其是 Aβ42)在大脑皮质的堆积是AD病理发生的早期触发因素[8-9],因此阻断或延迟AD早期Aβ的积聚成为治疗AD的切入点。在前期工作中,我们分别用VPA处理7月龄、9月龄APP23+/-单转基因小鼠,并进行了行为学、形态学和蛋白水平测定,VPA可显著改善AD模型鼠空间学习记忆的缺陷,可以减少脑内老年斑的形成[10],那么对于能更好的模拟AD疾病的APP/PS1双转基因模型鼠,VPA是否能发挥其神经保护功能呢?研究证明该模型鼠在4~5月龄时开始出现老年斑,因此我们选择4月龄老鼠在老年斑产生初期进行药物处理,随后进行了免疫组织化学和Thioflavin S染色检测,结果(图2)显示VPA组和对照组小鼠大脑皮质和海马内均出现了4G8阳性神经元及阳性斑块(老年斑),但VPA组小鼠皮质和海马等区域的老年斑数量显著少于对照组,且老年斑的体积也明显小于对照组。为验证老年斑数量减少是否源于Aβ水平降低,本实验ELASA结果显示,VPA处理组小鼠脑内Aβ40和Aβ42水平均有显著减少,其中以Aβ42减少更为明显,说明VPA是通过降低Aβ的生成来减少了老年斑的聚集。
过多的Aβ具有细胞毒性,引起神经细胞线粒体损伤,炎性反应导致神经细胞凋亡[11]。研究表明,Aβ的积聚能激活神经元细胞进入细胞周期并使细胞周期出现紊乱,从而也会引起细胞凋亡[9]。神经病理学发现,AD最早期的病理变化为海马内突触及可塑性受到损害、海马以及内嗅皮层出现神经元丢失。VPA既然可以减少Aβ的形成,那它能否防止神经元的丢失,在前期工作中,免疫荧光及银染结果显示,VPA注射组的APP23+/-小鼠海马及大脑皮质的神经元胞体及突起数量明显增多,且神经纤维排列有序。在本实验中,我们采用APP/PS1双转基因小鼠模型,通过Nissl染色发现VPA注射组小鼠的海马及大脑皮质区尤其是内嗅区的神经元数量明显增加(图3)。在AD发生的过程中,内嗅区的病变往往早于新皮质的变化。鉴于基底前脑、海马以及内嗅区的纤维联系,基底前脑-海马-内嗅区环路在AD发病中起重要作用。因此在内嗅区的神经元变化代表着AD的早期改变。为了进一步验证VPA所致的神经元增多是否由脑内神经元的凋亡减少引起,我们进行了TUNEI检测。TUNEL可标记组织细胞中核酸DNA片断的3′-OH末端,为检测DNA损伤的敏感方法,也是检测AD脑中细胞凋亡的有力证据。在我们的研究中,VPA组小鼠海马和皮质区TUNEL阳性细胞数生理盐水对照组明显减少,且相对着色浅,说明VPA的确减少了小鼠脑内神经细胞的凋亡(图4)。那么,其凋亡的相关机制又是什么,VPA所致的神经元数量增加的又一原因——VPA是否促进了小鼠脑内神经元的再生,关于这些问题有待进一步的研究。另外,tau蛋白过度磷酸化聚集形成的NFT是AD脑的另一病理特征,GSK-3β是引起tau蛋白磷酸化的主要激酶,那么,作为GSK-3β的抑制剂VPA,对于NFT又有什么影响,我们将继续深入研究。
[1]Muyllaert D,Kremer A,Jaworski T,et al.Glycogen synthase kinase-3beta,or a link between amyloid and tau pathology? [J].Genes Brain Behav,2008,7(S1):57-66.
[2]高宝兵,龙志敏,贺桂琼,等.常压高氧对APP/PS1双重转基因小鼠脑内老年斑及β-淀粉样蛋白的影响[J].中华神经科杂志,2010,43(3):222-226.
[3]Kockeritz L,Doble B,Patel S,et al.Glycogen synthase kinase-3—an overview of an over-achieving protein kinase[J].Curr Drug Targets,2006,7(11):1377-1388.
[4]Bhat RV,Budd Haeberlein SL,Avila J.Glycogen synthase kinase 3: a drug target for CNS therapies[J].Neurochem,2004,89(6):1313-1317.
[5]Hooper C,Killick R,Lovestone S.The GSK3 hypothesis of Alzheimer′s disease[J].J Neurochem,2008,104(6):1433-1439.
[6]Williams RS,Cheng L,Mudge AW,et al.A common mechanism of action for three mood-stabilizing drugs[J].Nature,2002,417(6886):292-295.
[7]Dickey CA,Loring JF,Montgomery J,et al.Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein+presenilin-1 transgenic mice[J].Neurosci,2003,23(12):5219-5226.
[8]Luque FA,Jaffe SL.The molecular and cellular pathogenesis of dementia of the Alzheimer′s type an overview [J].Int Rev Neurobiol,2009,84:151-165.
[9]陈祥,钟翎.阿尔茨海默病中β淀粉样蛋白神经毒性作用研究进展[J].中国神经精神疾病杂志,2008,34(5):315-317.
[10]Qing H,He G,Ly PT,et al.Valproic acid inhibits Abeta production,neuritic plaque formation,and behavioral deficits in Alzheimer′s disease mouse models[J].Exp Med,2008,205(12):2781-2789.
[11]Deshpande A,Mina E,Glabe C,et al.Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons[J].Neurosci,2006,26(22):6011-6018.
