非水毛细管电泳内标法测定麻黄中麻黄碱的含量
2011-07-27肖宇航荆照政
肖宇航 ,秦 群,荆照政
1.中南大学湘雅医院药剂科,湖南长沙 410008;2.中南大学湘雅医院中心实验室,湖南长沙 410008
麻黄碱(ephedrine)是中药麻黄的主要有效成分,具有松弛支气管平滑肌、扩张支气管、收缩血管、升高血压及兴奋中枢等作用。麻黄碱的测定方法很多,除容量分析法外,还包括薄层扫描法[1]、高效液相色谱法[2-4]、气相色谱法[5]、电位滴定法以及导数光谱[6]、双波长[7]等各种紫外分光光度法。目前高效毛细管电泳法已广泛应用于中药成分的分析[8],相比以上所述分析方法,它具有分析周期短,柱子不易污染,消耗试剂少,费用低的优点,特别适用于快速检验。目前也有毛细管电泳方法用于测定麻黄碱的报道[9-10]。本文采用非水毛细管电泳内标法定量测定麻黄碱,选择了合适的内标和非水电泳液,优选了仪器工作参数,简化了样本预处理操作,为麻黄碱的测定提供了一种简便、快速、定量可靠的方法,可用于医学研究样品和药材中麻黄碱的测定。
1 仪器与试药
System P/ACE 5010型毛细管电泳仪紫外检测器(Beckman 公司),未涂层熔融石英毛细管(47 cm×75 μm)(河北永年光导纤维厂),小型摇摆式中药粉碎机(浙江温岭市奥力中药机械有限公司),60目钢筛(浙江上虞五四纱筛厂),TGL16型台式高速冷冻离心沉淀机(湖南仪器仪表厂),VC130PB型超声液体破碎仪(美国SONICS&MATERIALS公司),WH-2微型旋涡混合仪(上海沪西分析仪器厂)。
麻黄碱标准品(SIGMA 公司,批号:Lot-17E6749),品红(上海沪峰生化试剂有限公司,批号:061018-2),草麻黄(山西大同),甲醇为色谱纯,其他试剂为分析纯。
2 方法与结果
2.1 溶液的配制
2.1.1 电泳缓冲液的制备 精密称取1.1402 g醋酸铵和1.2305 g醋酸钠,用甲醇溶解并精确定容至500 ml(pH 7.0),混匀后,4℃保存备用。
2.1.2 内标溶液的制备 精确称取10.0 mg品红,置于10 ml容量瓶内,用电泳缓冲液定容到10 ml(1 g/L),然后取0.5 ml置于10 ml容量瓶内,用电泳缓冲液定容到10 ml,混匀密封,得浓度为0.05 g/L的内标溶液,4℃保存备用。
2.1.3 对照品溶液的制备 精密称取盐酸麻黄碱标准物10.0 mg置于10 ml容量瓶内,用电泳缓冲液定容(浓度为1 g/L),混匀后取1 ml于5 ml玻璃容量瓶内,用电泳缓冲液定容到5 ml(浓度为 0.2 g/L),混匀密封,4℃保存备用。
2.1.4 样品溶液的制备 称取草麻黄干燥草质茎50 g于40℃烤12 h,然后粉碎成能过60目钢筛的粉末,称取100 mg粉末置于10 ml具塞玻璃管中,加入2 ml 1%HCl,40℃电热恒温水浴箱中浸泡12 h。每隔3 h超声提取1 min(勿使溶液发热),2000 r/min离心10 min,取1 ml上清液置1.5 ml EP管中,室温放置至样品液全干(约2 h)。精密量取1 ml电泳缓冲液置EP管内,并置于微型旋涡混合仪上使沉淀充分溶解后,12000 r/min离心10 min, 取100 μl上清液置于 0.5 ml塑料离心管内(麻黄碱含量大于0.2%的样品,需要酌情稀释样品液),加入10 μl内标溶液,混匀,装样进行电泳分析。
2.2 电泳条件
工作电压为20 kV,压力进样为10 Pa×10 s,电泳方向从正极到负极,柱温为25℃,检测波长为210 nm;运行缓冲液30 mmol/L醋酸铵和醋酸钠甲醇缓冲液(pH 7.0);两次运行之间,必需用0.1 mol/L的NaOH溶液冲洗柱子3 min,再用醋酸铵甲醇溶液冲洗柱子3 min。各类型非水溶液在使用之前均用0.45 μm微孔滤膜滤过。
在上述测定条件下,记录麻黄碱标准品和麻黄样品测定的NACE色谱图。见图1。
2.3 方法学考察
2.3.1 线性关系考察 取1 g/L麻黄碱对照品储备液适量,置于1.5 ml塑料离心管内,置于室温至样品液全干,用电泳缓冲液稀释,分别制成浓度为200.000、100.000、50.000、25.000、12.500、6.250和3.125 mg/L的对照品溶液。各取100 μl对照品溶液,分别加入10 μl内标溶液,混匀,进行标准曲线的测定。以标准物浓度(Y)对标准物峰高与内标物峰高比值(X)进行回归分析,得麻黄碱回归方程:Y=0.02775X+0.00129,相关系数r=0.9940。
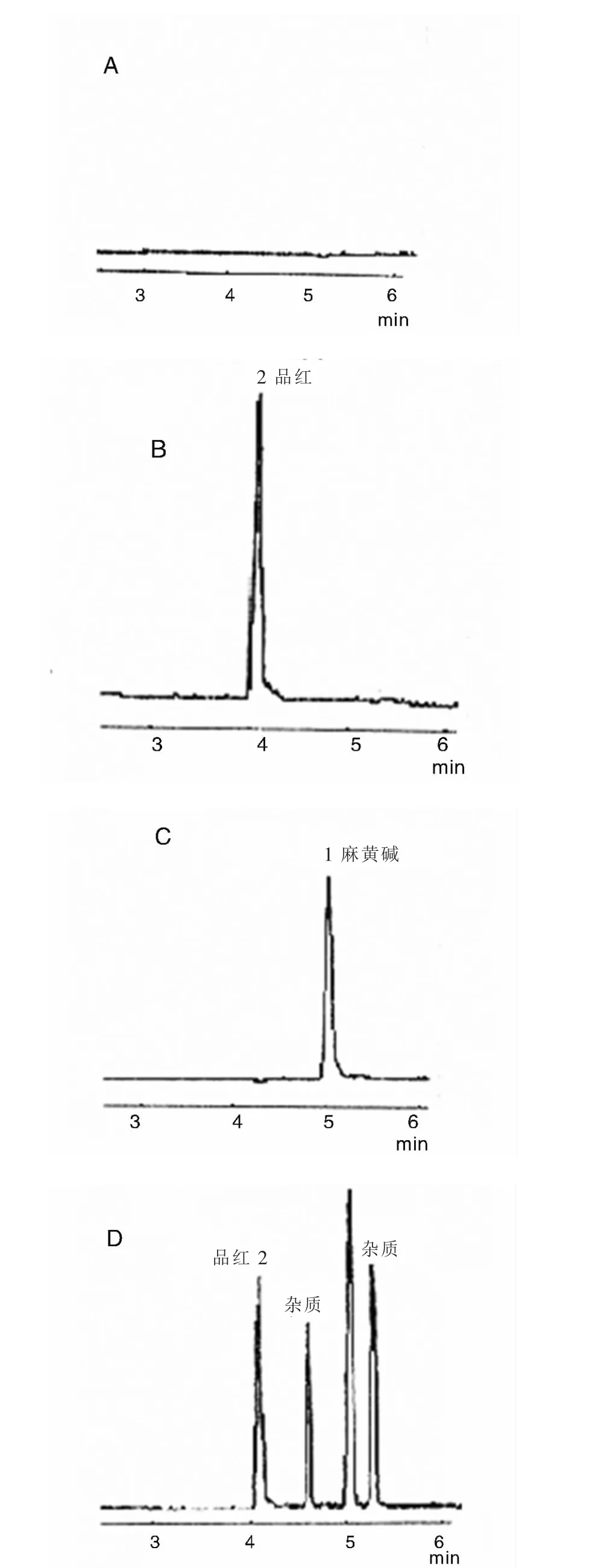
图1 NACE色谱图
2.3.2 最低检出浓度试验 取1 g/L的盐酸麻黄碱对照品溶液适量,置于1.5 ml塑料离心管内,置于室温至样品液全干,精密加入1 ml电泳缓冲液,置于微型旋涡混合仪上充分混匀,进样。当检测器显示的OD210值稳定后,启动电泳仪,在麻黄碱快去峰时,提前10 s随机记录麻黄碱在保留时间(tR)里的信噪吸收(absorbace,AU)最大值,按信噪比(S/N)约等于3,测得麻黄碱最低检出浓度为2 mg/L。
2.3.3 精密度试验 取0.1 g/L的麻黄碱对照品溶液100 μl,加入10 μl内标溶液,混匀,连续测定6次,测定结果显示RSD为2.05%,表明仪器精密度良好。选用1个样品,按样品处理程序进行操作,处理6份分别测定,测定结果显示,RSD为3.17%,表明本方法精密度良好。
2.3.4 加样回收率试验 分别选用含麻黄碱低和高的样品(含量约为0.1%和0.5%),每一个样品制成2份,每份取100 mg粉末置于10 ml玻璃管中,其中一份准确加入与样品含量相当的麻黄碱对照品(即第一组加入0.1 g/L麻黄碱1 μl,第二组加入0.5 g/L麻黄碱1 μl,),将其作为已知标准值的试验样品。而另一份样品作为空白对照样品。分别按样品处理程序进行操作,然后对4组单样进行测定,每份单样测定6次,结果显示本方法测定麻黄碱的回收率为97.47%~101.85%。测定结果见表1。
2.4 阴性空白试验
对麻黄碱的类似物鞣酸、黄酮苷、糊精和菊粉等按样品处理程序进行分析,未发现类似麻黄碱的未知物对毛细管区带电泳测定麻黄碱产生干扰。

表1 NACE测定麻黄碱回收试验结果
2.5 麻黄碱含量测定
麻黄碱含量以重量百分比表示,麻黄碱含量=(A/B)×F/W×100%。其中,A为麻黄碱峰高,B为内标物峰高,F为麻黄碱回归方程中的斜率,W为1 ml样品溶液里草麻黄的重量(mg)。经测定,草麻黄中麻黄碱含量为0.83%。
3 讨论
3.1 内标物的选择
非水毛细管电泳内标法测定麻黄碱的关键,在于选择合适的内标物质,使各种因素变化引起的误差最小。内标峰不仅要与所有样品峰分开,而且与被测样品峰靠得越近越好。本方法选择品红作为内标,初步达到优化内标物的要求。
3.2 电泳缓冲液的选择
用非水毛细管区带电泳测定麻黄碱时,用0.1 mol/L的NaOH溶液冲洗柱子,能显著提高灵敏度。在电泳缓冲液里加入醋酸钠,能使基线更平稳,另外电泳缓冲液里必须加入醋酸铵,否则检测不到样品峰。
3.3 电流的选择
在相同条件下,非水系统电流远小于水溶液系统,因此允许使用较高的运行电压和较大的电解质浓度,达到更加快速、高效分离的目的。但电泳时的电流过大会使基线不稳定,甚至于断电,可以稀释缓冲液使电流降低。本文中的分析电流控制在60 μA,能得到平稳的基线。
3.4 电压的选择
分离电压越高,样品的迁移时间越短,但分离效果不如电压较低时好。分离电压太低,样品的迁移时间过长,电泳峰随之展宽使得峰形劣化,定量误差加大。经过试验比较和优化,选择10 kV作为分离电压。
[1]徐先祥,孔树佳.薄层扫描法测定咳喘平颗粒中盐酸麻黄碱含量[J].中国医院药学杂志,2005,25(7):677-678.
[2]张涛.HPLC法测定复方苯海拉明麻黄碱糖浆中麻黄碱、苯海拉明的含量[J].药品鉴定,2009,6(21):42-44.
[3]陈小贞,陈小晖.HPLC法测定小儿咳喘灵口服液中盐酸麻黄碱的含量[J].中国现代医生,2008,46(7):105-106.
[4]宋志钊,陈昭,李星宇,等.高效液相色谱法测定滴通鼻炎水中盐酸麻黄碱的含量[J].广西医学,2008,30(4):548-549.
[5]顾金林,沈鸿.麻黄及其制剂中麻黄碱的含量测定[J].中国药师,2001,4(1):29-30.
[6]郭鸿宜,陈康.二阶导数光谱法测定不同产地草麻黄中总生物碱的含量[J].中药材,2004,27(10):738-739.
[7]朱雅艳,洪溪勇,华俊彦.双波长分光光度法测定氯麻滴鼻液中盐酸麻黄碱的含量[J].药品检测,2003,12(9):39.
[8]王永刚,孙学刚,魏凤环.葛根及粉葛化学成分谱HPCE方法学研究[J].南方医科大学学报,2008,28(8):1407-1408.
[9]钱燕娟,梁静,蒋小丰,等.高效毛细管电泳法测定麻黄汤中3种有效成分的含量[J].中国临床药学杂志,2008,17(6):371-373.
[10]刘长海,赵亮,张海,等.毛细管电泳法检查左旋麻黄碱中对映异构体杂质的研究[J].药学实践杂志,2009,27(1):33-37.
