蛋白酶抑制剂类抗癌药物的研究进展
2011-07-25杜登学王姗姗宫风华张志鹏
杜登学,王姗姗,宫风华,周 磊,张志鹏
(1.山东轻工业学院化学工程学院,山东 济南 250353;2.上海启达化工科技有限公司,上海 201512)
癌症的发病率近年来迅速增长,严重威胁着人类的生命健康,对癌症的治疗早已提上日程。癌症治疗药物种类繁多,近年来新兴的蛋白酶抑制剂类药物以其药效高、副作用小、协同作用好等优点受到重视。进入21世纪,蛋白酶抑制剂类抗癌药物的研究成为热点,较受关注的有硼替佐米(Bortezomib,1)、MG-132(2)、NLVS(3)、Tyropeptin A(4)、NPI-0052(5)、Lactacystin(6)、Omuralide(7)[1]等(结构如图1所示)。其中对硼替佐米的研究尤其活跃。

图1 蛋白酶抑制剂类抗癌药物的结构
1 简介
蛋白酶体是一个广泛分布于真核细胞质和细胞核中的高度保守的具有多种催化功能的蛋白酶复合物,尤其能催化水解连接有多聚泛素链的蛋白,是泛素—蛋白酶体(UPP)通路的主要组分。泛素-蛋白酶体类药是癌症治疗阵营中的新星,大量研究证实了其药效药性,拓宽了癌症治疗的途径。泛素-蛋白酶体调控着人体内必需、非必需或废弃的细胞蛋白的降解,该调控在许多癌症细胞中往往调控紊乱[2]。
治疗多发性骨髓瘤的二肽硼酸类化合物硼替佐米,在2003年得到了FDA的批准,率先在美国上市;2005年9月在我国上市,成为第一个上市的蛋白酶抑制剂类药物。随后一系列具有蛋白酶抑制活性的合成小分子化合物或提取的天然化合物进入临床试验。
2 结构机理
泛素-蛋白酶体是人体内降解蛋白质的主要途径(90%),其形状是由5个β-螺旋折叠形成一个中空并由一个α-螺旋填充的紧致球形[3],它的结构核心蛋白酶体(26S)是一个ATP依赖性蛋白水解酶复合体[4]。26S蛋白酶体主要由1个圆柱型的20S的催化中心和2个V型的19S的调节粒子组成。19S的调节粒子上有多个ATP酶活性位点,主要作用是识别已经泛素化的蛋白质,经过加工后,再将其转运至20S催化中心,赋予20S以ATP依赖性[5]。20S的催化中心主要由3个复合体酶的催化部位组成,分别为:类糜蛋白酶活性(ChTL)、类胰蛋白酶活性(TL)、蛋白酶样活性[6]。这些蛋白酶具有不同的活性,可以定向水解蛋白质上的特定氨基酸,达到水解蛋白质、释放多肽的目的。一般情况下蛋白酶抑制剂只抑制3个酶活性部位中的一个或两个来抑制蛋白质的水解,如硼替佐米只抑制类糜蛋白酶的活性[7]。
蛋白酶体的活性对细胞功能的维持非常重要。26S蛋白酶体对蛋白的降解依赖于对靶蛋白的泛素化和泛素化蛋白的识别,该过程由泛素激活酶E1、泛素结合酶E2和泛素连接酶E3依次完成[8~10]。蛋白酶体抑制剂能通过抑制蛋白酶体活性干扰和影响细胞正常的功能,通过调节蛋白质含量对肿瘤细胞的生长产生抑制作用。利用蛋白酶体抑制剂选择性抑制某些活性位点,从而改变蛋白酶体的酶切位点已成为免疫、抗炎等领域研究的热点。
3 抗癌特性
泛素-蛋白酶体主要有3个作用:加工非活性的蛋白前体成活性蛋白、调节体内蛋白平衡和识别除去异常蛋白[11,12]。作用于蛋白酶体的抑制剂的抗癌机理说法不一,但是在哺乳动物细胞的分裂增长中发挥着极为重要的作用是肯定的。临床研究表明,蛋白酶类抑制剂能够使细胞中的调节蛋白质集中在细胞质中,从而降低一些酶的活性,达到抑制恶性细胞分裂的目的[6]。有人指出因为肿瘤细胞中蛋白质的运转周期短,癌细胞对蛋白酶抑制剂更敏感[13];有人则认为在肿瘤细胞中,抑制剂与蛋白酶的作用时间比正常细胞长[14],阻碍癌细胞增长;还有人认为抑制剂在癌细胞中的降解速度较慢,作用时间长,从而达到治疗癌症的目的[6,15];肿瘤抑制因子在正常细胞中低水平表达,能很快被蛋白酶体降解,抑制剂能抑制酶体活性、增加肿瘤抑制因子浓度[16]。通过蛋白酶体来抑制蛋白质的降解是近年来癌症治疗的研究热点,它扩大了化学治疗药的靶向范围。
4 蛋白酶类抑制剂药物
4.1 醛基肽类
蛋白酶抑制剂的研究始于醛基肽类化合物,最早研究的主要有钙蛋白酶抑制剂Ⅰ(Ac-Leu-Leu-nLeu-al)和天然放射菌类产物亮太素(Ac-Leu-Leu-Arg-al)、MG-132、CEP-1612、CVT-634等。醛基肽类化合物一般具有强的缺电子羰基和弱的位阻,醛基与苏氨酸残基的-OH结合,形成一个半缩醛的结构,可逆性地抑制蛋白酶复合体的活性[17],但是醛基肽类抑制剂能很快被氧化成酸而被运出体外,天然化合物大多数作用强度较低、选择性不好,合成的稍好一些,但也未得到广泛的应用。后来人们为了增强其药性做了一些改进,比如在醛基的α位引入羰基、靶向载体[18]等,但总的来说应用不够广泛。
4.2 β-内酯类
β-内酯类化合物大多是从天然产物提取出来的。链霉菌代谢产物Lactacystin是从自然界分离出来的首个非肽类高度特异的蛋白酶抑制剂,Lactacystin在体内经过Jurkat T酶催化形成β-内酯活性中间体Omuralide(化合物7)[19],受丝氨酸残基上羟基的进攻,打开内酯环不可逆地抑制蛋白酶体的活性,其中酰胺环上的取代基对抑癌效果有很大的影响。
4.3 非共价类
一般的蛋白酶抑制剂都是以共价键同调节靶向结合,非共价抑制剂是同活性位点旁的特种基团发生非共价作用,例如氢键、范德华力等。该类化合物包括2-氨基苄基Staine结构为母体的化合物和环肽类化合物。TMC-95 A~D化合物是从菌属Apiosporamontagnei中提取的经过修饰的氨基酸残基环肽类化合物[20],该类化合物是天然提取产物中唯一的非共价抑制剂,药效在纳摩尔级。该类物质结构复杂,全合成有一定难度。
4.4 硼酸肽类
硼酸肽类化合物是现阶段最重要的蛋白酶抑制剂。一些研究已明确表明它有较好的蛋白酶体抑制活性和选择性,主要抑制核心蛋白酶体20S中类糜蛋白酶的活性。第一个硼酸肽类蛋白酶抑制剂药物硼替佐米是由日本武田和美国强生公司共同研发的,用于治疗复发和难治性多发性骨髓瘤[21]。
4.4.1 硼替佐米的结构性质
硼替佐米是哺乳动物细胞中26S蛋白酶体糜蛋白酶样活性的可逆抑制剂。活性中心是化合物中的硼酸结构,化合物中硼原子外层只有6个电子,含有一个空轨道,是一个典型的Lewis酸结构,可以与蛋白酶体中催化中心的丝氨酸残基中的-OH形成配位键,从而可逆性地抑制酶的活性。其结构改造主要通过改造二肽硼酸主链上的3个残基来进行。近年来用此法也发现了一些有较高医用性的药物,有的已进入临床前试验[22]。
体外研究表明,硼替佐米对多种类型的癌变细胞有细胞毒性。它在人体内可以引起肿瘤细胞凋亡,并增强肿瘤细胞对放疗和化疗的敏感性[23]。
多发性骨髓瘤被界定为不可治愈的疾病,临床表现不尽相同,硼替佐米对表达细胞周期蛋白D1的患者有更好的疗效。硼替佐米对多种肿瘤都有很好的治疗效果,如淋巴肿瘤、白血病、乳腺癌等,已经是抗癌领域的先锋药。对很多已经产生耐药性的肿瘤细胞有很好的抑制增殖作用。与其它抗癌药物如帕替尼、地塞米松、阿霉素、沙利度胺、马法兰等联合使用,具有疗效叠加及协同效应,还能延缓病变细胞的耐药性。
4.4.2 硼替佐米的合成
硼替佐米化学名称[(1R)-3-甲基-1-[[(2S)-1-氧-3-苯基-2-[(吡嗪羧基)氨基]丙基]氨基]丁基]硼酸,属于α-氨基硼酸肽类化合物,此类化合物的生物药效明显,但一般来说合成比较困难,主要原因是硼酸的α位存在一个不对称碳原子,它是化合物活性的重要组成部分。α-氨基硼酸部分的合成,以前基本上都是采取Matteson′合成法[24],由手性硼酸酯反应得α-氯硼酸酯,进一步转化成α-氨基硼酸。
此外,Sadighi′在Backer的研究基础[25]上设计了新的合成方法[26]:在金属铜配合物的催化作用下,亚胺中间体8与化合物9加成,生成α-氨基硼酸化合物10,这一步骤的优点是产率高、稳定性好、立体选择性也较好。然后在酸性条件下,选择性地水解氮端的磺酰基团,得到化合物11。化合物11在TBTU的催化下,与N-Boc-L-苯丙氨酸反应,经盐酸酸化得到化合物12。化合物12与(2-Pyrazine)COOH发生耦合反应,反应产物经水解硼酸保护基团后纯化即得到物硼替佐米(即化合物1)。合成路线见图2。
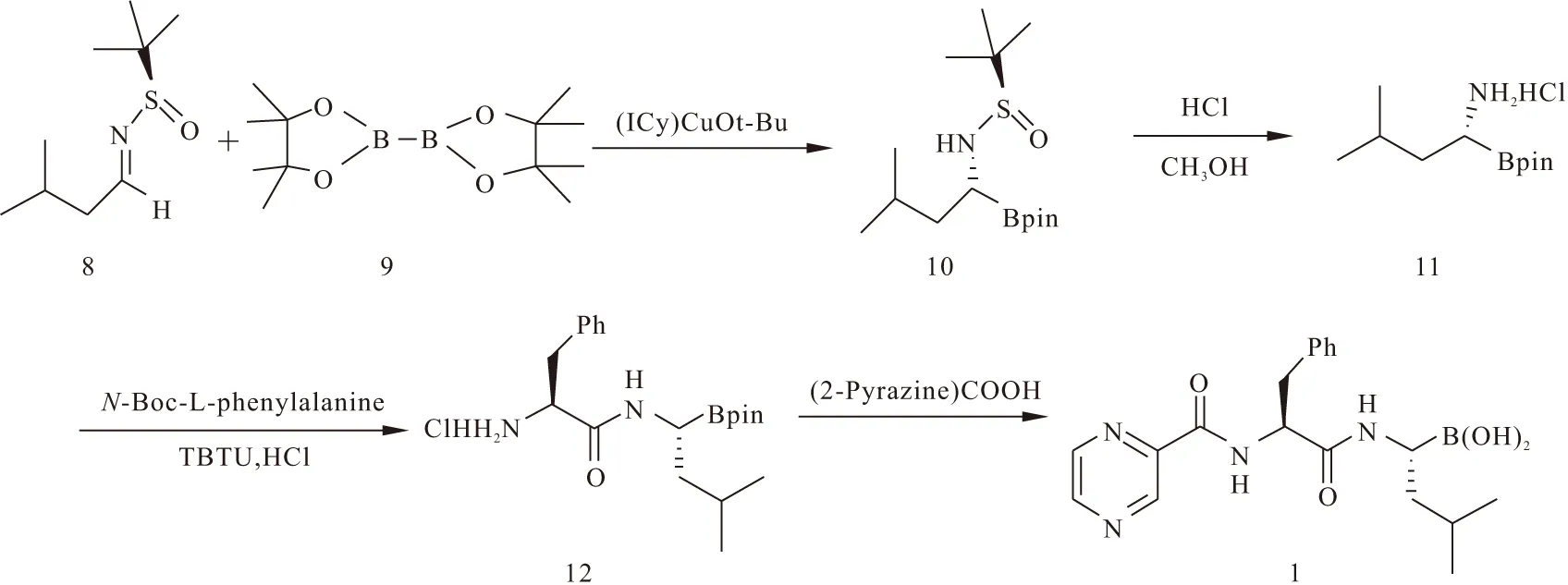
图2 硼替佐米的Sadighi′合成路线
Zhu等[1]以蒎烯和L-苯丙氨酸为原料合成硼替佐米。先将蒎烯氧化(该步需使用剧毒催化剂四氧化锇),再以二氯甲烷为起始反应物经酯化、取代、构型选择等得到重要的活性中间体16,再由化合物16和氨基酸缩合物17经缩合、硼酸化得目标产物,纯化后得到纯的硼替佐米,具体合成路线见图3。该合成路线比较长,多步反应需在较低温条件下才能完成,不太适合工业化生产。
除此之外还有多类抑制剂如乙烯基磺酰化合物、环氧酮类[27,28]、多酚类、β-内酰胺[29]、偶氮肽类等类型的抑制剂。
5 展望
当今虽然有很多种抗癌药物,但或多或少都存在一些问题,如药效差或毒副作用大等。泛素-蛋白酶体系统是哺乳动物细胞内主要的蛋白质控制系统,泛素-蛋白酶体监控体系参与细胞的整个增殖、分化和凋亡过程。异常的泛素-蛋白酶体行为与恶性肿瘤的发生和发展有着密切的关系,这主要是由于异常的泛素-蛋白酶体系统对细胞内重要功能蛋白质的降解失常所造成的。因此,研究靶向作用于蛋白酶体的药物对于控制治疗一些肿瘤疾病有重要的意义。上述的几类化合物中,有的已经得到了很好的应用,其中硼酸肽类和β-内酯类化合物对靶点的选择性好、副作用少,市场开发潜力较大。
[1] Zhu Y Q,Zhao X,Zhu X,et al.Design,synthesis,biological evaluation,and structure-activity relationship(SAR) discussion of dipeptidyl boronate proteasome inhibitors.Part I.Comprehensive understanding of the SAR ofα-amino acid boronates[J].J Med Chem,2009,52(14):4192-4199.
[2] Goldberg A L.Protein degradation and protection against misfolded or damaged proteins[J].Nature,2003,426(6968):895-899.
[3] Vierstra R D.Proteolysis in plants:Mechanisms and functions[J].Plant Mol Biol,1996,32(1-2):275-302.
[4] Hartmann-Petersen R,Seeger M,Gordon C.Transferring substrates to the 26S proteasome[J].Trends Biochem Sci,2003,28(1):26-31.
[5] Glickman M H.Getting in and out of the proteasome[J].Semin Cell Dev Biol,2000,11(3):149-158.
[6] Kisselev A F,Goldberg A L.Proteasome inhibitors:From research tools to drug candidates[J].Chem Biol,2001,8(8):739-758.
[7] DeMartino G N,Slaughter C A.The proteasome,a novel protease regulated by multiple mechanisms[J].Biol Chem,1999,274(32):22123-22126.
[8] Attaix D,Combaret L,Pouch M N, et al.Cellular control of ubiquitin-proteasome-dependent proteolysis[J].Anim Sci,2002,80(2):E56-E63.
[9] Pickart C M.Mechanisms underlying ubiquitination[J].Annu Rev Biochem,2001,70:503-533.
[10] Cardozo T,Pagano M.The SCF ubiquitin ligase:Insights into a molecular machine[J].Nat Rev Mol Cell Biol,2004,5(9):739-751.
[11] Karin M,Cao Y,Greten F R,et al.NF-κB in cancer:From inno-cent bystander to major culprit[J].Nat Rev Cancer,2002,2(4):301-310.
[12] Schubrt U,Anton L C,Gibbs J,et al.Rapid degradation of a large fraction of newly synthesized proteins by proteasomes[J],Nature,2000,404(6779):770-774.
[13] Lin Y J,Huang Y H,Zhen Y Z,et al.Rhein lysinate induces apoptosis in breast cancer SK-Br-3 cells by inhibiting HER-2 signal pathway[J].Acta Pharm Sin,2008,43(11):1099-1105.
[14] Masdehors P,Omura S,Merle-Béral H,et al.Increased sensitivity of CLL-derived lymphocytes to apoptotic death activation by the proteasome-specific inhibitor lactacystin[J].Br J Haematol,1999,105(3):752-757.
[15] Voorhees P M,Dees E C,O′Neil B,et al.The proteasome as a target for cancer therapy[J].Clin Cancer Res,2003,9(17):6316-6325.
[16] Adams J.The development of proteasome inhibitors as anticancer drugs[J].Cancer Cell,2004,5(5):417-421.
[17] Li B,Dou Q P.Bax degradation by the ubiquitin/proteasome-dependent pathway: Involvement in tumor survival and progression[J].Proc Natl Acad Sci,2000,97(8):3850-3855.
[18] Vivier M,Rapp M,Papon J,et al.Synthesis,radiosynthesis,and biological evaluation of new proteasonle inhibitors in a tumor targeting approach[J].J Med Chem,2008,51(4):1043-1047.
[19] Fenteany G,Standaert R F,Lane W S,et al.Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin[J].Science,1995,268(5211):726-731.
[20] Tsukamoto S,Yokosawa H.Natural products inhibiting the ubiquitin-proteasonle proteolytic pathway,a target for drug development[J].Curr Med Chem,2006,13(7):745-754.
[21] Adams J,Behnke M,Chen S,et al.Potent and selective inhibitors of the proteasome:Dipeptidyl boronic acids[J].Bioorg Med Chem Lett,1998,8(4):333-338.
[22] Piva R,Ruggeri B,Williams M,et al.CEP-18770:A novel,orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib[J].Blood,2008,111(5):2765-2775.
[23] Matteson D S,Sadhu K M,Lienhard G E.(R)-1-Acetamido-2-phenylethaneboronic acid.A specific transition-state analog for chymotrypsin[J].J Am Chem Soc,1981,103(17):5241-5242.
[24] Papandreou C N,Logothetis C J.Bortezomib as a potential treatment for prostate cancer[J].Cancer Res,2004,64(15):5036-5043.
[25] Mann G,John K D,Baker R T.Platinum-catalyzed diboration using a commercially available catalyst:Diboration of aldimines toα-aminoboronate esters[J].Org Lett,2000,2(14):2105-2108.
[26] Beenen M A,AN C,Ellman J A.Asymmetric copper-catalyzed synthesis ofα-amino boronate esters fromN-tert-butanesulfinyl aldimines[J].J Am Chem Soc,2008,130(22):6910-6911.
[27] Rydzewski R M,Burrill L,Mendonca R,et al.Optimization of subsite binding to theβ5 subunit of the human 20S proteasome using vinyl sulfones and 2-keto-1,3,4-oxadiazoles:Synthesis and cellular properties of potent,selective proteasome inhibitors[J].J Med Chem,2006,49(10):2953-2968.
[28] Marastoni M,Baldisserotto A,Cellini S,et al.Peptidyl vinyl ester derivatives:New class of selective inhibitors of proteasome trypsin-like activity[J].J Med Chem,2005,48(15):5038-5042.
[29] Imbach P,Lang M,Garcia-Echeverria C,et al.Novelβ-lactam derivatives:Potent and selective inhibitors of the chymotrypsin-like activity of the human 20S proteasome[J].Bioorg Med Chem Lett,2007,17(2):358-362.
