牛黄化毒片的质量标准研究*
2011-06-21苗淑杰
刘 冰,王 佳,苗淑杰
( 1.天津市药品检验所,天津 300070; 2.天津同仁堂集团股份有限公司,天津 300385 )
牛黄化毒片收载于《卫生部药品标准中药成方制剂第二十册》,执行标准为WS3-B-3567-98。该药是由天南星(制)、连翘、金银花、白芷、甘草、乳香、没药、人工牛黄8味药材组成,有解毒消肿,散结止痛的功效,可以用于疮疡、乳痈、红肿疼痛的治疗。现执行的质量标准仅规定了性状及绿原酸的薄层色谱鉴别和检查项,而其中白芷具有祛风湿,活血排脓,生肌止痛的功效。为有效地控制药品质量,本文增加了连翘、甘草和人工牛黄的薄层鉴别方法,及白芷中欧前胡素的含量测定方法。修订后的标准可用于牛黄化毒片的质量控制。
1 仪器与试药
Agilent 1100高效液相色谱仪(包括在线脱气机、四元泵、自动进样系统、DAD检测器、Chem Stations色谱工作站)。连翘对照药材(批号120908-200613)、甘草对照药材(批号120904-200512)、胆酸对照品(批号100078-200414) 、猪去氧胆酸对照品(批号100087-200610)和欧前胡素对照品(批号为110826-200712,供含量测定用,含量99.9%)均由中国药品生物制品检定所提供。乙腈为色谱纯,水为去离子水。高效硅胶G薄层板(山东烟台化学工业研究所,批号070316)。牛黄化毒片样品(天津市同仁堂股份有限公司提供,批号为584004、783001、788002、889002)。
2 薄层色谱鉴别
2.1连翘的鉴别
2.1.1溶液制备
2.1.1.1供试品溶液的制备 分别取本品6片,除去包衣,研细,加甲醇15 ml,超声处理15 min,滤过,滤液作为供试品溶液。
2.1.1.2对照药材溶液的制备 连翘对照药材0.5 g,加甲醇10 ml,加热回流20 min,滤过,滤液作为对照药材溶液。
2.1.1.3阴性对照溶液的制备 按处方配比(连翘除外) 根据制法制成的阴性样品,取阴性样品粉末1.8 g,加甲醇15 ml,照“2.1.1.1”项下方法提取,作为阴性对照溶液。
2.1.2薄层条件与结果 照薄层色谱法(《中国药典)2005年版一部附录Ⅵ B)试验,吸取供试品溶液、对照药材溶液及阴性对照溶液各6~8 μl ,分别点于同一高效硅胶G薄层板上,以三氯甲烷-甲醇(5∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105 ℃加热至斑点清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性对照溶液无干扰。见图1。
2.2甘草的鉴别
2.2.1溶液制备
2.2.1.1供试品溶液的制备 分别取本品16片,除去包衣,研细,加甲醇20 ml超声30 min,滤过,滤液挥干,加水20 ml溶解,滤过,滤液加乙酸乙酯萃取2次,每次15 ml,挥干乙酸乙酯层,残渣加甲醇1 ml溶解,作为供试品溶液。
2.2.1.2对照药材溶液的制备 甘草对照药材1 g,同法制成对照药材溶液。
2.2.1.3阴性对照溶液的制备 按处方配比(甘草除外) 根据制法制成的阴性样品,取阴性样品粉末4.8 g,加甲醇20 ml,照“2.2.1.1”项下方法提取,作为阴性对照溶液。
2.2.2薄层条件与结果 照薄层色谱法(《中国药典》2005年版一部附录Ⅵ B)试验,吸取供试品溶液、对照药材溶液及阴性对照溶液各4~6 μl ,分别点于同一高效硅胶G薄层板上,以甲苯-乙酸乙酯-甲酸(10∶1∶0.5)的上层溶液为展开剂,展开,取出,晾干。再重新配置展开剂进行二次展开,喷以10%硫酸乙醇溶液,在105 ℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,分别显相同颜色的荧光斑点,阴性对照溶液无干扰。见图2。
2.3人工牛黄的鉴别
2.3.1溶液制备
2.3.1.1供试品溶液的制备 分别取本品8片,除去包衣,研细,加甲醇10 ml,超声处理5 min,滤过,作为供试品溶液。
2.3.1.2对照药材溶液的制备 胆酸、猪去氧胆酸对照品,分别加甲醇制成每1 ml含1 mg的溶液,滤液作为对照品溶液。
2.3.1.3阴性对照溶液的制备 按处方配比(人工牛黄除外) 根据制法制成的阴性样品,取阴性样品粉末2.4 g,加甲醇10 ml,照“2.3.1.1”项下方法提取,作为阴性对照溶液。
2.3.2薄层条件与结果 照薄层色谱法(《中国药典》)2005年版一部附录Ⅵ B)试验,吸取供试品溶液、对照药材溶液及阴性对照溶液各4~6 μl ,分别点于同一高效硅胶G薄层板上,以正己烷-醋酸乙酯-醋酸-甲醇(6∶32∶1∶1)为展开剂,展开,取出,晾干,喷以10%磷钼酸酸乙醇溶液,在105 ℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,分别显相同颜色的斑点,阴性对照溶液无干扰。见图3。
3 含量测定
3.1色谱条件 参照《中国药典》[1]2005年版一部有关测定方法,确定检测波长为300 nm。Agilent zorbax 色谱柱C18(250 mm×4.6 mm, 5 μm);乙腈-水(52∶48)为流动相;柱温30 ℃;流速1.0 ml/min,进样量为20 μl。上述色谱条件下,理论板数按欧前胡素峰计算应不低于5000;分离度应大于1.5。
3.2溶液的制备
3.2.1对照品溶液 取欧前胡素对照品适量,精密称定,加甲醇制成每1 ml含0.01 mg的溶液,即得。
3.2.2供试品溶液的制备 取本品10片,除去包衣,精密称定,研细,取约1 g,精密称定,置具塞锥形瓶中,精密加入甲醇溶液25 ml,称定重量,超声处理60 min,放冷,再称定重量,用甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,即得。
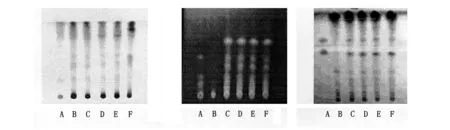
A 对照药材 A 对照药材 A 对照药材
B 供试品584004 B 阴性对照 B 阴性对照
C 供试品783001 C 供试品584004 C 供试品584004
D 供试品788002 D 供试品783001 D 供试品783001
E 供试品889002 E 供试品788002 E 供试品788002
F 阴性对照 F 供试品889002 F 供试品889002
图1连翘TLC色谱图图2甘草TLC色谱图图3人工牛黄TLC色谱图
3.2.3阴性样品溶液的制备 按处方配比,取除白芷外的其他药材,按牛黄化毒片的工艺方法,制得阴性样品,再按供试品溶液的制备方法制成阴性样品溶液。精密吸取对照品、供试品和阴性样品溶液各20 μl,注入液相色谱仪。在与欧前胡素对照品色谱峰相对应的保留时间处,无色谱峰出现,阴性样品溶液无干扰。见图4。
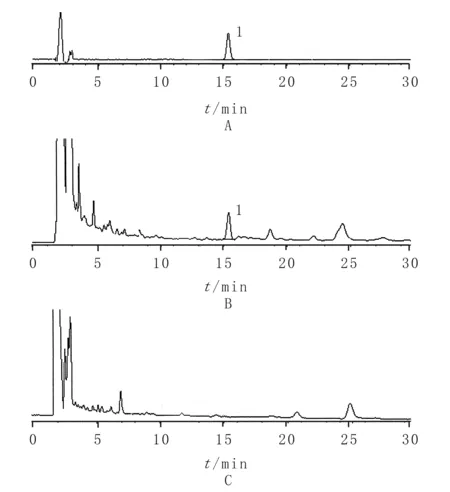
1.欧前胡素
3.3线性关系考察 取欧前胡素对照品,精密称定,加甲醇制成每1 ml含0.100 4 mg的溶液,作为储备液。分别精密量取储备液1、3、5、10和15 ml,置50 ml量瓶中,加甲醇稀释至刻度,摇匀,精密吸取上述溶液及储备液20 μl,注入液相色谱仪,以浓度(mg/ml)为横坐标,峰面积为纵坐标,绘制标准曲线。得标准曲线方程:Y=4.932 5×104X-4.967 4(r=0.999 9)。结果表明,欧前胡素在0.002 008~0.100 4 mg/ml范围内线性关系良好。
3.4进样精密度试验 取批号为889002的样品1份,按“3.2.2供试品溶液的制备”项下方法操作,按“3.1 ”项下色谱条件分析,精密吸取供试品溶液20 μl,连续进样6次,测定样品中欧前胡素峰面积值,平均峰面积值为466.458 068,RSD为0.53%(n=6)。结果表明,精密度符合要求。
3.5重现性试验 取批号为889002的样品6份,按“3.2.2”项下供试品溶液的制备法操作,按“3.1” 项下色谱条件分析,测定欧前胡素的含量,结果测得样品平均含量为0.072 3 mg/片,RSD为0.63 %,结果表明,重现性符合要求。
3.6稳定性试验 取批号为889002的样品1份,按“3.2.2供试品溶液的制备”项下方法操作,按“3.1” 项下色谱条件分析,分别精密吸取供试品溶液20 μl,分别于0、2、5.5、9、16和24 h进样,测定欧前胡素的含量RSD为0.77%,结果表明,供试品溶液在24 h内测定稳定。
3.7回收率测定 取批号为889002的样品6份,各0.5 g,精密称定,精密加入每1 ml含欧前胡素对照品0.004 823 mg的甲醇溶液25 ml,称定重量,超声处理60 min,放冷,再称定重量,用甲醇补足减失的重量,滤过,取续滤液,作为供回收率测定用的供试品溶液,按“3.1”项下色谱条件分析,计算回收率。结果平均回收率为98.77%,RSD为0.67%。
3.8样品测定 取其他牛黄化毒片样品3批,按“3.2.2”项下供试品溶液的制备方法操作,按“3.1 ”项下色谱条件分析测定,计算3批样品含量,结果见表1。

表1 样品测定(n=2)
4 讨论
4.1检测波长选择 取欧前胡素对照品溶液,在200~400 nm波长处进行扫描,结果表明:欧前胡素对照品在249 nm与300 nm波长处有较大吸收峰。参照《中国药典》2005年版一部“白芷”项下含量测定,以300 nm波长作为含量测定波长。
4.2提取溶剂及提取方法的选择 取批号为889002的样品10片,除去包衣,精密称定,研细,取约1 g,精密称定,以甲醇、乙醚为溶剂,分别采用超声处理、加热回流及索氏提取3种方法进行提取,按“3.1”项下色谱条件分析。结果表明采用甲醇作为溶剂,3种提取方法测定结果基本一致,明显高于以乙醚为溶剂时3种提取方法测定结果。故选取甲醇作为溶剂,超声处理的提取方法。
4.3提取时间的考察 取批号为889002的样品10片,除去包衣,精密称定,研细,取约1 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 ml,称定重量,分别超声处理30、60、90和120 min,放冷,再称定重量,用甲醇补足减失的重量,滤过,取续滤液作为供试品溶液。按“3.1”项下色谱条件分析。结果显示4个提取时间测定结果基本一致,为保证提取完全,故采用60 min作为提取时间。
4.4溶剂用量的考察 取批号为889002的样品10片,除去包衣,精密称定,研细,取约1 g,精密称定,置具塞锥形瓶中,精密加入甲醇50 ml,称定重量,超声处理60 min,放冷,再称定重量,用甲醇补足减失的重量,滤过,取续滤液作为供试品溶液。按“3.1”项下色谱条件分析。结果显示2个提取溶剂用量含量测定结果无明显差异,故选择25 ml作为牛黄化毒片的提取溶剂用量。
4.5不同色谱柱含量测定的影响 取批号为788002样品溶液,使用Agilent 1100高效液相色谱仪,分别使用Dikma C18(4.6 mm×250 mm,5 μm)色谱柱和phenomenex C18(250 mm×4.6 mm, 5 μm) 色谱柱,按照“3.1”色谱条件分析,测定样品中欧前胡素的含量,结果三种色谱柱测得的含量分别为0.064 6、0.065 6和0.064 4 mg/片。三者含量平均值为0.064 9 mg/片,对测定结果无明显的影响。
4.6其他
中药成分复杂,鉴别仅以单一成分为对照,难以反映药品质量,在尽可能的情况下,应以有效成分和对照药材为对照,并尽量简化操作过程。连翘中有效成分为连翘苷,实验中发现,按《中国药典》2005年一部中连翘药材的方法提取、分离,结果色谱斑点拖尾严重[2],互相掩盖,难以达到鉴别效果。而本实验可使供试品色谱的分离效果达到最佳,具有较强的专属性。
在甘草中有效成分为甘草酸铵,实验中发现,其内所含成分较为复杂,不易于分离,根据文献[3]试过多种提取方法如正丁醇萃取、乙醚萃取等,但其效果不显著,色谱斑点仍拖尾严重、互相掩盖,不能达到鉴别分离的效果。而本实验可使供试品色谱的分离效果达到最佳,具有较强的专属性。
1 中国药典.一部.2005:4,59,68,117,136
2 王春红.补血胶囊的TLC定性鉴别.湖北中医杂志,2002,24(4):51
3 韩桂茹,徐韧柳,胡盂魁. 利肺片质量标准研究.中国现代应用药学杂志,2004,21(3):209
