反相高效液相色谱法测定醋酸甲萘氢醌片的含量*
2011-04-09龚士学程辉跃
林 昀,龚士学,程辉跃
(重庆市药品检验所,重庆 401121)
醋酸甲萘氢醌又名维生素K4,是维生素类药,主要适用于肠道吸收不良及各种原因所致阻塞性黄疸、慢性溃疡性结肠炎等肠道吸收功能减低等造成维生素K缺乏的凝血障碍性疾病。现行标准中,醋酸甲萘氢醌片的含量测定方法为紫外分光光度法[1-3]。笔者按质量标准方法验证指导原则[1],建立了测定醋酸甲萘氢醌片含量的反相高效液相色谱法,现报道如下。
1 仪器与试药
Agilent1100型高效液相色谱仪(配有紫外检测器和二极管阵列检测器);AgilentC18柱(250mm×4.6mm,5μm),AichromBond-AQC18柱(250mm×4.6mm,5μm),WelchMaterialsC18柱(150mm× 4.6mm,5μm);AE 240型电子天平(瑞士梅特勒)。醋酸甲萘氢醌对照品(中国药品生物制品检定所,批号为0228-9802);样品(3个厂家共6批,均为市售品);乙腈为色谱纯,水为二次蒸馏水。
2 方法与结果
2.1 色谱条件选择
2.1.1 测定波长选择
取醋酸甲萘氢醌对照品约10 mg,用适量流动相溶解并稀释成每1 mL中含0.02 mg的溶液,照紫外-可见分光光度法[2005年版《中国药典(二部)》附录ⅣA],在230~400 nm波长范围内扫描(图1)。结果285 nm波长处有最大吸收,故以此为测定波长。
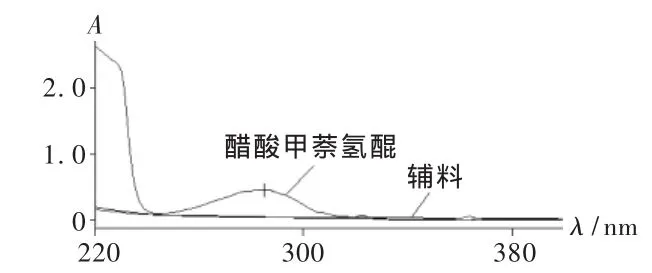
图1 醋酸甲萘氢醌紫外扫描图谱
2.1.2 流动相选择
由于国内外药典均未收载该品种的高效液相测定方法。笔者经考察和筛选,最终决定以乙腈-水(65∶35)为流动相,柱温为室温,检测波长285 nm,流速1.0mL/min,进样量20μL。在此色谱条件下,被测物质峰与相邻杂质峰分离度良好,保留时间适中,峰纯度因子为999.0,色谱图见图2 C。

图2 高效液相色谱图
2.1.3 空白辅料影响及破坏性试验
称取按处方量配制的空白辅料(相当于1片的量),加流动相约100mL,置37℃水浴中,超声处理10min,放冷,滤过,作为空白辅料溶液。取该续滤液按上述色谱条件测定,结果见图2 B。另取本品细粉适量(约相当于醋酸甲萘氢醌20mg),分别经碱破坏、酸破坏、氧化破坏、光照破坏及热破坏后用流动相稀释成质量浓度为0.02 g/L的溶液,照上述色谱条件进样分析,在3倍主峰保留时间内,未见破坏试验产生的各杂质峰,证明本实验色谱条件可行。
2.2 溶液制备
精密称取样品(批号为080201)20片,研磨后取6份(每份相当于醋酸甲萘氢醌10mg),分别置50mL量瓶中,用流动相溶解并稀释至刻度,摇匀,滤过,精密量取5mL,置50mL量瓶中,用流动相稀释至刻度,摇匀,作为供试品溶液;另取醋酸甲萘氢醌对照品适量,同法配制成对照品溶液。
2.3 系统耐用性试验
分别用 Agilent C18柱(250 mm×4.6 mm,5μm)、Aichrom-Bond-AQ C18柱(250 mm×4.6 mm,5μm)、Welch Materials C18柱(150mm×4.6mm,5μm)3种色谱柱按上述条件试验,结果基本一致,说明本方法的耐用性较好。可见,辅料在醋酸甲萘氢醌峰的保留时间处无干扰,对测定无影响。
2.4 方法学考察
线性范围考察与定量限确定:精密称取醋酸甲萘氢醌对照品39.71mg,置100mL量瓶中,用适量流动相溶解并稀释至刻度,摇匀,精密量取1.0,2.5,5.0,10.0,15.0mL,分别置100mL量瓶中,加流动相稀释至刻度,摇匀,制成质量浓度分别为4.0,10.0,20.0,40.0,60.0μg/mL的对照品溶液,进样测定,记录色谱图,以质量浓度 X对峰面积 Y进行回归,得回归方程 Y=27 001X+2.943 7,r=1.000(n=5)。结果表明,醋酸甲萘氢醌质量浓度在 4.0~60.0μg/mL范围内与峰面积线性关系良好。以 S/N=10计算,本品的定量限确定为54.21 ng/mL。
精密度试验:取质量浓度为20.0μg/mL的对照品溶液,照上述色谱条件进样分析,连续5次,记录色谱图。结果峰面积的 RSD为0.04%(n=5)。
稳定性试验:取质量浓度为20.0μg/mL的供试品溶液,分别在0,1,2,4,6h时测定。结果峰面积的 RSD为0.21%(n=5),表明醋酸甲萘氢醌溶液在室温下6h内稳定。
重复性试验:取同一批样品,照上述色谱条件依法测定。结果醋酸甲萘氢醌片的平均含量为98.79%,RSD为0.91%(n=6),表明方法重复性良好。
回收率试验:分别取对照品约16,20,24 mg各3份,精密称定,分别置50mL量瓶中,加入相应辅料,用流动相适量溶解并稀释至刻度,摇匀,滤过,作为对照品溶液。精密量取续滤液5.0mL,置100mL量瓶中,加流动相稀释至刻度,摇匀,作为供试品溶液。精密量取供试品溶液和对照品溶液各20μL,依法测定,计算回收率。结果见表1。
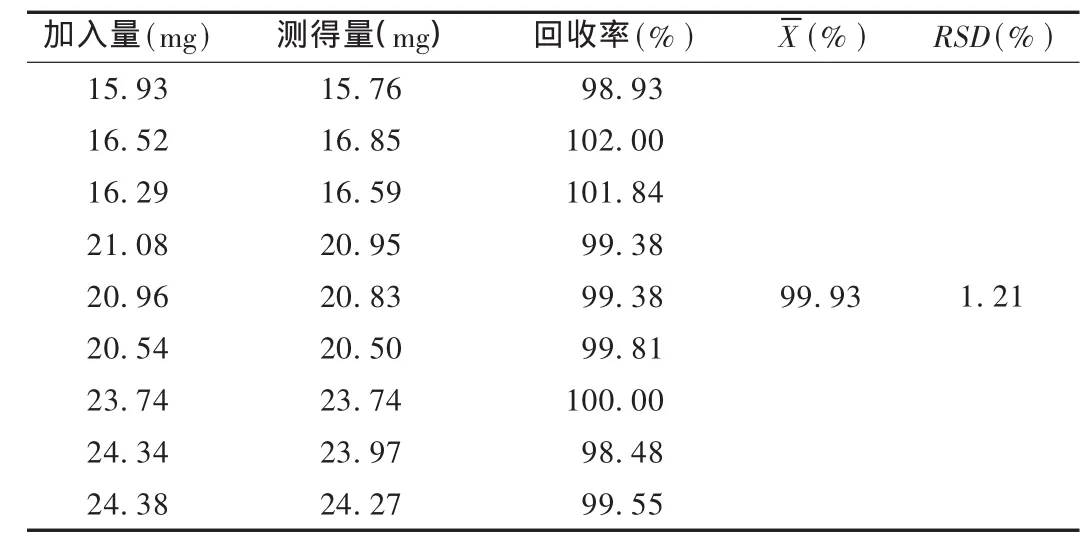
表1 醋酸甲萘氢醌回收率试验结果(n=9)
2.5 样品含量测定
取3个厂家共6批样品各2份,按本试验所用方法与2005年版药典方法分别测定含量。结果见表2。
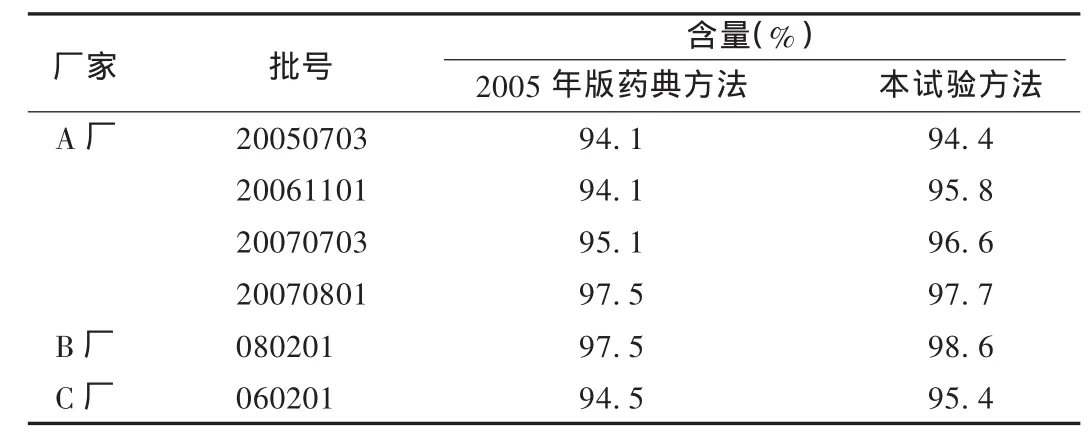
表2 样品含量测定结果
3 讨论
现行质量标准中的紫外分光光度法专属性不强,易受溶剂和辅料的影响。虽有人采用高效液相色谱法测定含量[4],但测定波长接近末端吸收,在200~400 nm波长范围内进行全波长扫描,醋酸甲萘氢醌在末端比杂质有更大的吸收,影响其他杂质的检测,而在285 nm波长处各杂质峰均有不同程度的吸收,利于主成分的含量测定,故将检测波长定在285 nm。
在流动相选择过程中,乙腈的量过少时,分析时间较长,且峰形不好,当乙腈∶水(65∶35)时,主峰与各杂质峰能很好分离,分离度大于1.5,且峰形良好。
由含量测定结果可以看出,两种方法的测定结果差异不大。但用高效液相色谱法可以有效地将主成分与杂质分开,该方法专属性强、灵敏度高、重复性好、操作简便、结果准确可靠,适用于醋酸甲萘氢醌制剂的质量控制。
[1]国家药典委员会.中华人民共和国药典(二部)[M].北京:化学工业出版社,2005:840,附录172-173.
[2]顾弟元,陈佩绵.紫外分光光度法测定维生素Kd片剂含量方法的改进[J].苏州医学院学报,1995,15(2):372-373.
[3]郭金鹏,王英英.分光光度法测定片剂中维生素K4的含量[J].河北医学学院学报,1989,10(3):157-158.
[4]陈建琴,郑 洁.高效液相色谱法测定醋酸甲萘氢醌片的含量[J].中国医院药学杂志,2005,25(4):376-377.
