应用反相高效液相色谱定性和定量浮游植物光合色素
2011-03-14黄邦钦
胡 俊,柳 欣,王 磊,黄邦钦
(1.厦门大学 福建省海洋环境科学重点实验室 海洋与环境学院,福建 厦门 361005; 2.华东师范大学 河口海岸学国家重点实验室,上海 200062)
由于叶绿素、类胡萝卜素等光合色素具备:(1)只有营光合作用的浮游生物细胞中含有; (2)一些光合色素仅出现于某一类浮游生物; (3)光合色素分子不稳定,细胞死亡后会迅速分解; (4)不同的光合色素对可见光的吸收光谱特征不同。因此,光合色素的测定不仅可以用于指示浮游植物的总生物量,还可作为生物标志物,用于指示浮游植物的类群组成、生理状况[1]、水团的特征[2,3]、真光层中的输出生产力[4]、水质状况[5]及浮游植物原位生长速率等[6],已经被国内外海洋生态学家广泛接受。随着矩阵因子化程序CHEMTAX[7]的提出,进一步推动了光合色素在海洋生态学研究中的应用。
海水中光合色素的提取、分离、定性和定量分析是应用光合色素法准确评价水体中浮游植物生物量及类群组成的前提和基础。但由于光合色素种类繁多,分子结构差异不大,且对光、温、酸碱、氧十分敏感,个别光合色素还会形成同分异构体,特定海区特定光合色素浓度较低等特点,给光合色素的提取、分离、定性与定量过程带来较大困难。目前反相高校液相色谱法是国内外光合色素的分离、分析的首推方法。借助该方法,国内研究者已能将大量的叶绿素及类胡萝卜素进行很好的分离[8-9]。陈纪新等[10]借助二极管阵列检测器(DVD),选择合理的洗脱程序,能将叶绿素a(Chla)和二乙烯基叶绿素a(DV-Chla)较好地分离。而姚鹏等[11]和朱卓毅[12]在Zapata等[13]方法基础上做一定修改后,同样获得了Chl a和DV-Chl a较好的分离效果。但目前在光合色素的定性、定量方面的专门研究还较少,实际分析工作中常会遇到不少问题,比如光合色素浓度与色谱峰面积的转换系数(fp)不准确,将无法准确估算水体中光合色素的实际浓度,也影响文献数据之间的可比性。作者购买通过藻类单种纯培养获得的高纯度光合色素标准品(丹麦 DHI-14C公司),采用 Furuya等[14]的提取方法、在Balow等[15]和陈纪新等[16]的分离方法基础上做一定修改后,分析测定了19种关键的浮游植物光合色素的保留时间、吸收光谱、最大吸收波长(λmax)、校正曲线及响应因子(fp)等参数,并对影响各参数的因素进行讨论与分析,为国内同行在相关领域的研究提供借鉴与参考。
1 材料和方法
1.1 仪器与材料
仪器:Agilent l100 Series液相色谱工作站,结合二极管阵列检测器(DAD),Eclipse XDB C8色谱柱(Agilent,Germany)。
材料:光合色素标准品,从丹麦DHI-14C公司购买,光合色素标准品的名称(中文名称、英文名称及缩写)、初始浓度及溶解体系如表1所示。

表1 光合色素标准品的名称、缩写、浓度及溶解体系Tab.1 The names,abbreviations,concentrations and solvents of the photosynthetic pigments
1.2 方法
1.2.1 洗脱程序
光合色素的高效液相色谱分析流动相由A和B组成,其中 A 为V甲醇:V1M乙酸铵=4:1,B 为 100%甲醇。洗脱程序如表2所示。

表2 光合色素HPLC分离分析梯度洗脱程序Tab.2 The linear gradient elution protocol of HPLC-based photosynthetic pigment analysis
1.2.2 响应因子测定
将购得的光合色素标准品进行梯度稀释(至少4个浓度梯度),测定不同浓度的谱峰面积(检测波长为 440 nm),以光合色素浓度Cp作为 y轴,峰面积 Ap作为x轴,以光合色素浓度和峰面积作散点图,通过线性回归得出线性方程(1),其斜率即为响应因子(fp)。

1.2.3 定量
各光合色素标准品在不同稀释浓度下进行色谱分析,测得不同浓度下各色素在440 nm波长的峰面积。利用校正因子fp,根据公式(2)进行光合色素浓度的计算:

式中,Cs为光合色素质量浓度,单位为ng/L;Ap各光合色素洗脱峰的面积,单位为mAU*s;fp为各光合色素浓度与峰面积线性回归方程的斜率,单位为mg/mAU;vext为进行色素抽提时所用提取液体的体积,单位为10-3L;vfilt为采样时过滤海水的体积,单位为10-3L;vinj为高效液相色谱分析时的进样体积,单位为10-3L; B为缓冲液的稀释因子。
2 结果
2.1 光合色素标准品纯度的检验
标准光合色素在运输、保存过程中会因出现不同程度的降解而纯度下降。因此,标准品使用前必须检验其纯度。标准品的纯度检验通过 Chem-station程序完成。具体操作是分段检测光合色素标准品在440 nm的吸收峰峰宽范围内的吸收光谱特征,结果显示,各光合色素标准品在峰宽范围内各时间点的吸收光谱重叠较好,表明标准品纯度高。(图 1a为BUT440 nm的吸收峰,图1b为BUT在吸收峰的范围内分段检测其吸收光谱)。

图1 光合色素19-丁酰基氧化岩藻黄素的纯度检验Fig.1 Confirmation of the peak purity on the example of 19’-butanoyloxy -fucoxanthin
2.2 光合色素的液相色谱分离
混合标准品的色谱分析情况如图2所示。在本实验体系下各种标准品的保留时间均小于 32 min,表明该方法有较高的样品分析速率。另外,该方法能将DV-Chla和Chla,LUT和ZEA,Chlb和DV-Chlb等极性差异不大的光合色素较好地分离,但是Chlc1和Chlc2仍然是在同一个峰中被洗脱出来。

图2 反相高效液相色谱对混合标准光合色素的色谱分析图Fig.2 RP-HPLC chromatograms of mixed photosynthetic pigment standards
2.3 光合色素的吸收谱峰
在本实验体系下,叶绿素类及类胡萝卜素类在300~800 nm波长范围内的吸收光谱特征如图3及图4所示。叶绿素类均存在两个较强吸收峰,其一位于蓝光区(300~500 nm),另一个区域在红光区(550~700 nm)。Chla及DV-Chla(图3e、f)在两个区域的最大吸收波长处的吸收强度相当,Chlc(图3a、b)在红光区最大吸收波段的吸收强度则明显弱于蓝光区。Chlb及DV-Chlb(图3c、d)在410 nm处存在吸收的相对低值。因此,根据吸收光谱特征上的差异完全可以将Chla、b和c区分开来。
虽然Chla与DV-Chla吸收光谱的形状非常相似,但它们之间也存在差异(图3e、f)。Chla在蓝光区的最大吸收波长为432 nm,而DV-Chla的最大吸收波长红移了 10 nm,为 442 nm。类似的情况在Chlb与DV-Chlb之间也存在,DV-Chlb的最大吸收波长红移了8 nm。因此,虽然Chla/b与DV-Chla/b的保留时间非常接近,凭此差异也完全可以将二者区分。

图3 叶绿素类光合色素的光吸收光谱特征Fig.3 Absorption spectra of Chlorophylls
类胡萝卜素对可见光的吸收主要在蓝光区,波长范围在 350~550 nm。在此范围内,一般情况下均有 2~3个吸收峰(图 4d、g),但是会因溶剂不同而有所差异。若以Ⅰ、Ⅱ和Ⅲ表示波长由短至长的三个吸收峰,Ⅰ和Ⅲ通常会因发色基团的干扰而形成肩峰,如DIAD、ALL和DIAT等,Ⅰ已经形成了肩峰,Ⅲ保留较好(图4h、i、j)。个别光合色素的Ⅰ和Ⅲ也会被Ⅱ完全覆盖而形成单峰特征,如 PER和CANT(如图 4a、m)。
与叶绿素类相比,类胡萝卜素类之间的吸收光谱差异较小,有些光合色素最大吸收波长仅差 1~2 nm。如,PERI和CAN,只有Ⅱ出现,且只相差1 nm。而DIAD、ALL和DIAT的Ⅰ均为肩峰,Ⅱ的波长相同,只有Ⅲ有微小的差别。因此,仅凭Ⅰ、Ⅱ和Ⅲ的差异很难将其区分; 而不同溶剂对Ⅰ、Ⅱ和Ⅲ的波长也会有影响,这进一步加大了它们之间的定性难度。因此,在定性过程中必须参考光合色素的保留时间。
2.4 光合色素的定量
各已知浓度的色素标准品经过一系列稀释,并与测得的440 nm波长的峰面积进行线性回归得出的校正工作曲线如图 5所示,响应系数(fp),保留时间及其最大吸收波长如表3所示。
由表可知,不同的光合色素的响应系数差别较大。Chla、Chlb、DV-Chla和PRA的fp值较高,分别为0.417,0.505,0.276和0.25。表明该类光合色素的仪器信号较弱; 而 Chlc2,FUCO,VIO,DIAD,NEO,ALL,DIAT,ZEA以及β-CAR等fp值较低,其值均低于 0.1; 其他光合色素的fp值介于 0.2和 0.1之间。
3 讨论
3.1 光合色素的定性
3.1.1 保留时间

图4 利用反相高效液相色谱测得类胡萝卜素的光吸收光谱特征Fig.4 Absorption spectra of carotenoids obtained
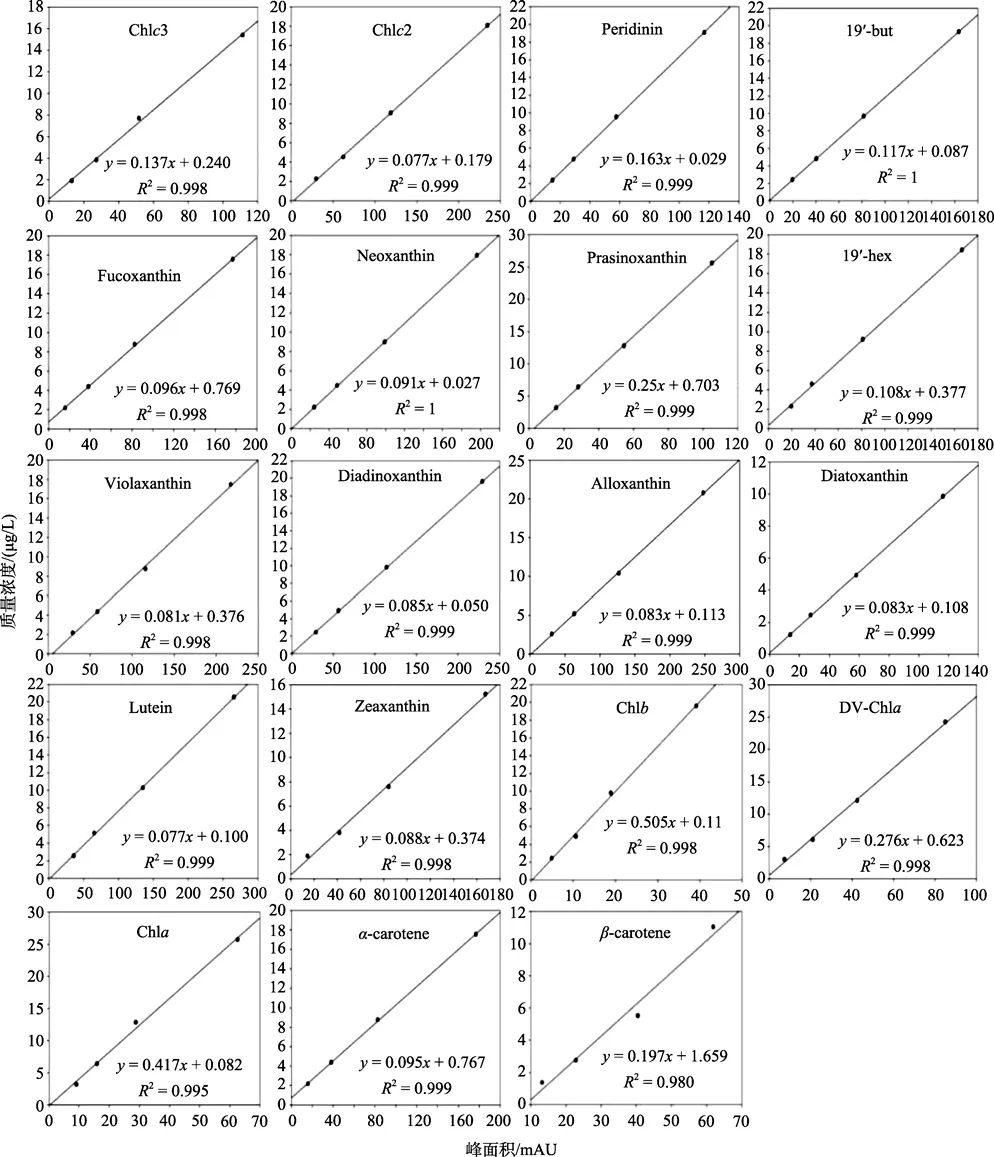
图5 标准光合色素的回归曲线Fig.5 Calibration curves for the photosynthetic pigment standards
保留时间是对光合色素进行定性的一个重要参数。在一定的体系中各光合色素的保留时间是相对稳定的。但是在进行样品分析时,通常会发生保留时间漂移现象。此现象的发生可能是环境温度、流动相、色谱柱性质发生改变产生导致,如流动相脱气过程会改变流动相的气压平衡,而且一轮梯度洗脱后固定相极性是否回到初始状况等均会使保留时间改变。因此,进行样品分析前应先进甲醇空白样分析,以保证样品分析前流动相气压达到平衡。另外,将分离柱放置在柱恒温器或水浴中,可达到稳定分析柱温度(变化范围<0.1℃),有利于改善保留时间漂移现 象。
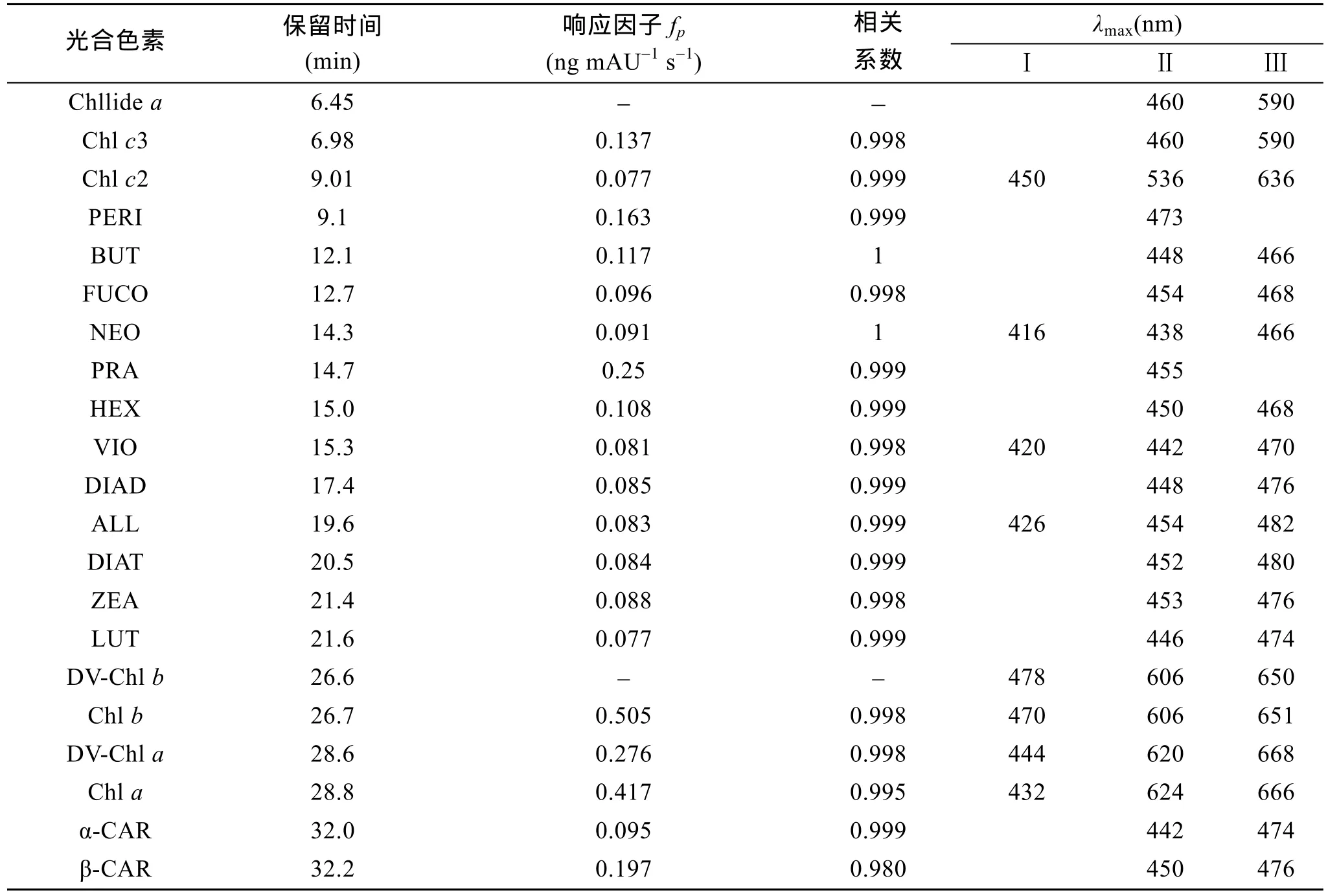
表3 光合色素标准品的保留时间、响应因子(fp)、相关系数及最大吸收波长Tab.3 Retention times,response factors,important coefficients and wavelengths of the absorption maxima of pigments in RP-HPLC analysis
3.1.2 出峰顺序
出峰顺序是由各光合色素的相对极性所决定,与提取剂、流动相的组成及分析柱的选择紧密相关,但在一定的体系下是相对稳定的。本研究中HEX的保留时间短于VIO,因而先被洗脱出峰。而在某些方法中,如Van等[17]的方法中,二者的出峰顺序与本研究刚好相反。在Stón等[18]的方法中,HEX的极性变化更大,其峰先于NEO。此外,本研究中ZEA和LUT两峰的先后顺序也与Stón等[18]恰好相反。因此在对以上这几个光合色素的定性时必须结合其保留时间和吸收光谱来加以判断。另外,光合色素的吸收光谱特征也会随分析体系的差异而有所不同。因此,最佳的定性方法是在光合色素标准品的辅助下,获得自己的分析体系下各种光合色素的保留时间以及吸收光谱特征。
3.2 光合色素的定量
3.2.1 光合色素标准品的获得
光合色素标准品的获得有两种途径,其一,从化学药品公司购买,如Chla、Chlb和 β-CAR可从Sigma-Aldrich公司购得,ZEA和LUT可以从Roth化学试剂公司购买。但由于商业途径提供的光合色素标准品的种类有限,往往难以满足对海洋中主要光合色素的定性、定量需求。虽然丹麦 DHI-14C公司提供了种类较全的光合色素标准品,但其量少(2.5 mL),浓度也不够高,使得样品中的光合色素浓度落在工作曲线之外。另外,该标准品一旦拆封后就得立即使用。若拆封后需要保存一段时间后继续使用,则必须确定标准品是否降解,并采用分光光度法重新标定浓度。相对其高昂的价格,这不是经济的选择。其二,利用藻类的纯种培养自己制备。利用制备或半制备型 HPLC将需要的光合色素分离纯化、收集后用分光光度法,根据其最大波长处的消光系数进行浓度标定,浓度计算如公式(3)所示:

其中Cd为光合色素质量浓度,单位为mg/L,Amax是在最大吸收波长下的吸光值,A750nm表示在750nm处的吸光值,l为比色皿的宽度,E为光合色素在最大吸收波长下的消光系数[18,19],该值如表4所示。

表4 在一定溶剂下各光合色素在最大吸收波长处的消光系数Tab.4 Extinction coefficients of pigments at the maximum absorb wavelengths
但是这个过程较为繁琐,且必须具备含有目标光合色素的藻株。有些特征光合色素含有的藻株在实验室内较难培养。部分藻株需从美国 Provasoli-Guillard国家海洋藻类培养中心(CCMP)购买。
3.2.2 响应因子(fp)
响应因子是色谱光电信号转化成光合色素浓度的关键系数,不同光合色素的响应因子不同。将本研究的fp值与文献值[20]线性相关性分析,得到线性方程为y=1.045x,R2值为 0.957,且斜率接近 1(图 6)。但各点的分布虽然靠回归曲线很近,但并未完全落于线上,表明二者之间仍然有差异,这可能是实验体系差异导致。Stoń等[18]比较了 LiChroCART Hypersil ODS 和 LiChroCART LiChrospher 100 RP1两款不同色谱柱在相同的流动相及柱温条件下各种光合色素的响应因子的差异,发现响应因子受固定相的影响较大,且变化规律不同。如,Chlb,采用不同的分离柱测得其fp值分别为0.427 5和0.600 9,之间相差将近50%; 本研究所得的fp值介于二者之间,为0.505。但采用不同的分离柱测得的Chla的fp值差别不大,分别为0.445 8和0.414 7; 本研究中Chla的fp为0.417。若使用不同的fp值计算同一个样品时,会使得Chlb的浓度差别较大,而Chla浓度变化不大。而PRAS与Chlb情况类似。因此,在选用不同类型色谱柱或更换新的色谱柱时,必须重新测定fp值。另外,高效液相色谱的光源更换、仪器搬运也可能会导致电信号的波动,fp会发生变化,也需对其重新测定。
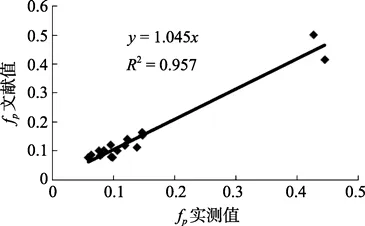
图6 fp实测值与文献值之间的的相关性分析Fig.6 Relationship between the fp tested and the fp from references
4 小结
利用本实验室建立的RP-HPLC实验体系,分析了19种浮游植物常见光合色素标准品的保留时间、吸收光谱、最大吸收波长、校正曲线及响应因子等参数。结果显示,在该实验体系下,19种光合色素都能够较好地分离和检测,但HEX与NEO、PRA之间及LUT和ZEA之间的出峰顺序与文献报道的其他实验体系的结果有所差异。因此,对以上光合色素进行定性时,不但要参考色素的出峰顺序、保留时间,还必须同时参考色谱峰的吸收光谱特征。另外,通过将本研究获得的光合色素的响应因子(fp)与文献值进行比较,作者发现不同光合色素的响应因子在不同的实验体系下的变化趋势不同。因此,当分析实验体系或仪器状态发生改变时,不能继续沿用原先的fp值来计算光合色素的浓度,而应当利用光合色素标准品对fp进行重新测定。
[1]Guillard R R L,Murphy L S,Foss P,et al.Synechococcusspp.as likely zeaxanthin-dominant ultraphytoplankton in the North Atlantic [J].Limnol And Oceanogr,1985,30(2):412-414
[2]Lohrenz S E,Carroll C L,Weidemann A D,et al.Variations in phytoplankton pigments,size structure and community composition related to wind forcing and water mass properties on the North Carolina inner shelf [J].Cont Shelf Res 2003,23:1 447-1 464.
[3]Hassen B M,Drira Z,Hamza A,et al.Phytoplankton dynamics related to water mass properties in the Gulf of Gabes:Ecological implications [J],J Mar System,2009,75:16-226.
[4]Claustre H.The trophic status of various oceanic provinces as revealed by phytoplankton pigment signatures[J].Limnol and Oceanogr,1994,39(5):1206-1210.
[5]Sherrard N J,Nimmo M,Llewellyn C A.Combining HPLC pigment markers and ecological similarity indices to assess phytoplankton community structure:An environmental tool for eutrophication? [J].Science of the Total Environment,2006,361:97-110.
[6]Pinckney J L,Richardsona T L,Millieand D F,et al.Application of photopigment biomarkers for quantifying microalgal community composition and in situ growth rates [J].Organic Geochemistry,2001,32:585-595.
[7]Mackey M D,Mackey D J,Higgins H W.CHEMTAX a program for estimating class abundances from chemical markers:application to HPLC measurements of phytoplankton [J].Mar Ecol Prog Ser,1996,144:265-283.
[8]王海黎,洪华生,徐立.利用反相高效液相色谱技术分离和测定海洋浮游植物叶绿素和类胡萝卜素[J].海洋学报,1999,4:6-9.
[9]彭兴跃,洪海征,黄明,等.利用光合色素研究厦门海域超微型浮游植物群落结构[J].台湾海峡,2002,21(1):78-85.
[10]陈纪新,叶翔,陈小鹏,等.有机试剂提取浮游植物光合色素的研究[J].厦门大学学报(自然科学版),2005,44(1):102-106.
[11]Yao P,Yu Z G,Deng C M.Pigment signatures of some diatoms isolated from China seas[J].Acta Oceanologic Sinica,2006,25(1):108-118
[12]朱卓毅.长江口及邻近海域低氧现象的探讨[D].上海:华东师范大学,2007:24-55.
[13]Zapata M,Rodríguez F,Garrido J L.Separation of chlorophylls and carotenoids from marine phytoplank-ton:a new HPLC method using a reversed phase C8 column and pyridine containing mobile phases[J].Mar Ecol Prog Ser,2000,195:29-45.
[14]Furuya K,Hayashi M,Yabushita Y.HPLC Determination of Phytoplankton Pigments Using N,N-Dimethyl form amide[J].J Oceanogr,1998,54:199-203.
[15]Barlow R G,Cummings D G,Gibb S W.Improved resolution of mono and divinyl chlorophylls a and b and zeaxanthin and lutein in phytoplankton extracts using reverse phase C-8 HPLC[J].Mar Ecol Prog Ser,1997,161:303-307.
[16]陈纪新,黄邦钦,刘媛,等.应用特征光合色素研究东海和南海北部浮游植物的群落结构[J].地球科学进展,2006,21(7):738-746.
[17]Van Heukelem L,Thomas C S.Computer-assisted high-performance liquid chromatography method development with applications to the isolation and analysis of phytoplankton pigments [J].J Chromatogr A,2001,910:31-49.
[18]Stoń-Egiert J,Kosakowska A.RH-HPLC determination of phytoplankton pigments - comparison of calibration results for two columns [J],Mar Biol,2005,147 (1):251-260.
[19]Wright S W,Jeffrey S W,Mantoura R F C.Evaluation of method s and solvents for pigment extraction in phytoplankton pigments in ceanography.Monographs on Oceanographic MethodologY[M].Paris:UNESCO,1997:261-282.
[20]Stoń-Egiert J,Kosakowska A.Phytoplankton pigments designation - an application of RP-HPLC in qualitative and quantitative analysis [J].J App Phycol,2002,14(3):205-210.
