β-环糊精硝基衍生物HPLC柱分离手性药物扑尔敏
2011-02-03沈静茹韦慧慰余学红朱财延
沈静茹,韦慧慰,余学红,朱财延,毛 康
(1中南民族大学化学与材料科学学院,分析化学国家民委重点实验室,武汉430074;2中南民族大学校医院,武汉430074)
药物扑尔敏大多以消旋体的形式出售,故扑尔敏(结构式见图1)的手性分离分析及制备研究对控制药品质量和各对映体潜在副作用的探究具有重要意义.目前,HPLC中扑尔敏手性分离主要通过营造手性环境对其分离,如:采用羟丙基-β-环糊精作为流动相添加剂,使得扑尔敏两异构体达到基线分离,分离度Rs>2.99[1].但此法所消耗手性选择剂量远大于手性固定相法;通过右旋扑尔敏的分子印记聚合物作为固定相,在正相模式下使扑尔敏达到部分分离[2];采用β-环糊精手性柱,质谱检测、分离人体血液中扑尔敏及其主要代谢物,最佳条件下扑尔敏两异构体之间分离度Rs达到1.17[3]等;通过原子转移自由基聚合制成固定相,在反相模式下手性分离血液中的扑尔敏及其主要代谢物[4].上述手性固定相法均未使扑尔敏两异构体达到基线分离,本文通过实验室自制的双[-6-氧-(-3-间硝基苯磺酰基-丁二酸-1,4 单酯)-4-]-β-环糊精键合硅胶固定相HPLC柱,分别在正相和反相模式下分离扑尔敏消旋体,正相模式分离条件下最佳分离度Rs达到1.37;而反相模式下达到了基线分离,最佳分离度Rs为10.58.通过精密度试验及加标回收实验考察分离分析方法.为进一步制成制备柱,实现扑尔敏单一对映体的分离制备提供了条件.

图1 扑尔敏结构式Fig.1 Structure of chlorpheniramine
1 主要仪器及试剂
1.1 试剂
异丙醇(色谱纯,天津市科密殴化学试剂有限公司)、正己烷(分析纯,天津市科密欧化学试剂有限公司)、三乙胺(分析纯,上海金山亭化工试剂厂)、乙酸(分析纯,国药集团化学试剂有限公司)、双[-6-氧-(-3-间硝基苯磺酰基-丁二酸-1,4 单酯)-4-]-β-环糊精(β-CD-M2)(实验室自制)、超纯水(美国 Moleculer超纯水机生产)、马来酸氯苯那敏标样(含量99.7%,中国药品生物制品检定所)、马来酸氯苯那敏片(湖北华中药业有限公司).
1.2 仪器
P230-Ⅱ高效液相色谱仪(大连依利特分析仪器有限公司)、pHS-3C型酸度计(上海伟业仪器厂)、0.22 μm微孔滤膜(上海市新亚净化器件厂)、双[-6-氧-(-3-间硝基苯磺酰基-丁二酸-1,4 单酯)-4-]-β-环糊精键合硅胶固定相HPLC柱(β-CD-M2键合柱)(实验室自制)[5].
1.3 色谱条件
色谱柱:β-CD-M2键合柱 4.6 mm ×150 mm(实验室自制);流动相:正己烷-异丙醇、异丙醇-0.3%乙酸三乙胺(TEAA)缓冲溶液;检测波长:254 nm;柱温:25℃;流速:正相体系为0.5 mL/min,反相体系为0.2 mL/min.
2 结果与讨论
2.1 正相模式下手性分离扑尔敏
在β-CD-M2键合柱上,以正己烷-异丙醇为流动相,在异丙醇体积分数为30% ~60%变化范围内对扑尔敏消旋体进行手性分离,结果见表1.由表1可见,随着流动相中异丙醇体积分数增加,保留时间减短.其主要原因为异丙醇增强了流动相的极性,使溶剂与扑尔敏的相互作用增强,进而增强了流动相的洗脱强度,当异丙醇体积分数为35% ~55%时扑尔敏两手性异构体保留时间有差异,实现了部分分离,最佳分离度Rs为1.37.
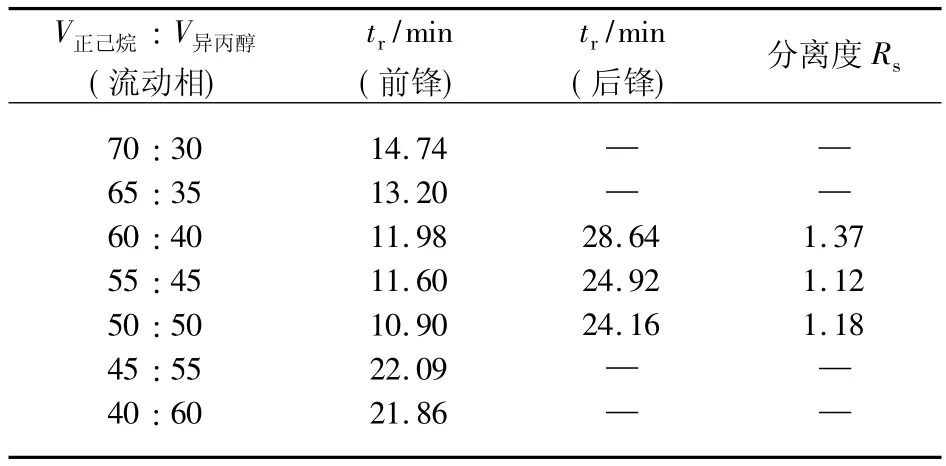
表1 正己烷-异丙醇为流动相手性分离结果Tab.1 Result of chiral separation using hexane-isopropyl alcohol asmobile phase
选择流动相配比(V正己烷∶V异丙醇)分别为60%∶40%和55%∶45%时,所得的拆分结果见图2.不同流动相配比中,两对映体的色谱峰(曲线a)均未被溶剂峰(曲线b)干扰.
2.2 反相模式下手性分离扑尔敏
2.2.1 流动相异丙醇-0.3%TEAA配比对扑尔敏手性分离的影响
在β-CD-M2键合柱上,以异丙醇-0.3%TEAA(pH4.5)为流动相,在异丙醇体积分数为40% ~90%变化范围内对扑尔敏消旋体进行手性分离,结果见表2.随着流动相中TEAA体积分数的增大,分离度呈上升趋势;超过40%,分离度变化不明显,而后峰的出峰时间延长.故在保证分离的情况下,为减短分析时间,选择VTEAA∶V异丙醇=40%∶60%为最佳配比.
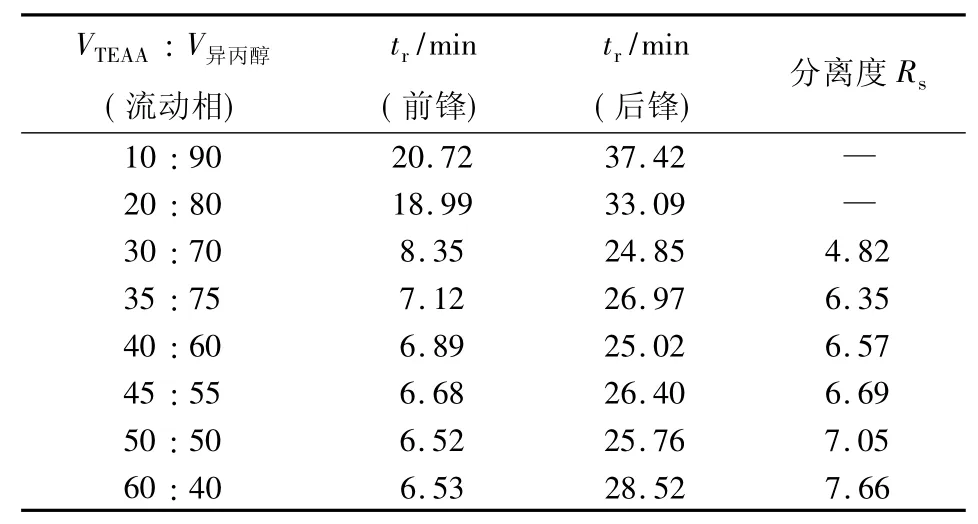
表2 流动相中异丙醇-0.3%TEAA配比的影响Tab.2 Effect of the ratio of0.3%TEAA and isopropyl alcohol in themobile phase
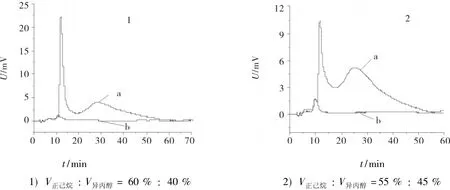
图2 正相模式下扑尔敏的手性分离Fig.2 Chiral separation of chlorpheniramine at normal phasemodel
选择流动相异丙醇 -0.3%TEAA(pH4.5),其体积配比为60%∶40%时所得的拆分结果见图3.
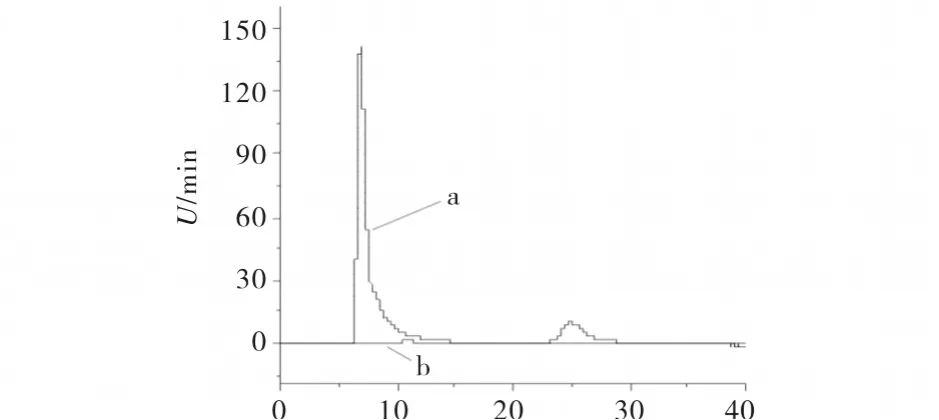
图3 溶剂为异丙醇-TEAA时扑尔敏在流动相中的手性分离Fig.3 Chiral separation of chlorpheniramine using isopropyl alcohol-TEAA in themobile phase
2.2.2 流动相中缓冲液pH值对扑尔敏手性分离的影响
在 β-CD-M2键合柱上,以V异丙醇∶V0.3%TEAA=60%:40%为流动相,在缓冲液 pH分别为2.57、3.10、3.55、4.52、5.13、5.90、6.78 时进行扑尔敏的手性分离,结果见图4.
由图4可知,随着酸度的增加,分离度呈上升趋势,pH 3.1 时最大,Rs=10.58;pH >3.0,分离度有所下降,但均Rs>6.00,达到基线分离.扑尔敏两对映体与固定相两者均因有苯环而可形成π-π共轭,均含有易产生氢键的元素(N、O元素等),且扑尔敏pKa=9.2[6],达到基线分离 pH 值范围(2.5 ~6.7)扑尔敏两异构体带电,故两对映体与固定相易形成分子间作用力(如氢键或静电作用等),且因作用力强弱不同,导致两对映体与固定相的结合力有所差异,因而保留时间相距甚远,分离度大.
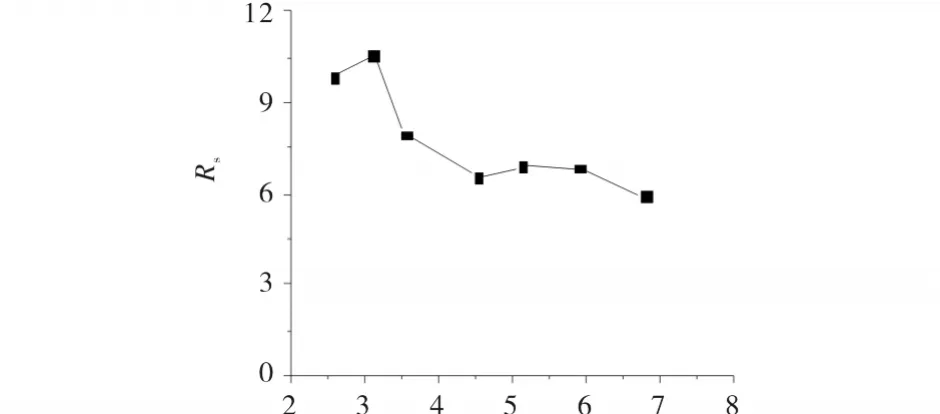
图4 缓冲液酸度对扑尔敏手性分离的影响Fig.4 Effect of buffer acidity
2.3 方法评价
2.3.1 考察分离的线性范围
配置扑尔敏标准品(99.7%)溶液,浓度分别为:0.055、0.137、0.273、0.342、0.687、1.367 mg/mL.以β-CD-M2键合硅胶为固定相,室温流动相流速0.2 mL/min,进样量20μL,测定扑尔敏分离的线性范围,分别考察了峰高-浓度、峰面积-浓度标准曲线.前锋峰高-浓度标准曲线方程为Y=1124.7691X,线性相关系数r=0.9985,峰面积-浓度标准曲线方程为Y=84258.63998X,线性相关系数r=0.9998;后峰峰高-浓度标准曲线方程为Y=1122.59395X,线性相关系数r=0.9985,峰面积-浓度标准曲线方程为Y=84237.81861X,线性相关系数r=0.9998.
2.3.2 精密度考察
在β-CD-M2键合硅胶为固定相,室温,流速0.2 mL/min,进样量20 μL 色谱条件下测定 0.137 mg/mL扑尔敏中2组份的精密度,进样次数n=12,得精密度考察结果:前峰峰高相对标准偏差为3.77%,峰面积相对标准偏差为1.05%;后峰峰高相对标准偏差为 2.90%,峰面积相对标准偏差为3.15%.
2.3.3 加标回收试验
色谱条件:β-CD-M2键合硅胶为固定相,室温,流速0.2 mL/min,进样量20 μL.在0.20 mg/mL 扑尔敏样品中分别测定加入浓度为 0.16、0.20、0.24 mg/mL扑尔敏标准品,计算加标回收率.测定次数n=4,测定结果见表3.

表3 扑尔敏加标回收率(n=4)Tab.3 Percent recovery of chlorpheniramine
由表3可知,加标浓度为0.16 mg/mL时,以峰高-浓度标准曲线计算回收率为86.14% ~90.68%,以峰面积-浓度标准曲线计算回收率为90.90%~95.77%;加标浓度为0.20 mg/mL 时,以峰高-浓度标准曲线计算回收率为94.69% ~106.93%,以峰面积-浓度标准曲线计算回收率为86.36% ~89.14%;加标浓度为 0.24 mg/mL 时,以峰高-浓度标准曲线计算回收率为105.41% ~108.76%,以峰面积-浓度标准曲线计算回收率为 89.15%~91.01%.
说明加标浓度与样品浓度相当时用峰高定量此法的准确度较高(回收率为94.69% ~106.93%),低于或高于样品浓度20%测定值会略有偏低或偏高的现象.
3 结语
以实验室自制的双[-6-氧-(-3-间硝基苯磺酰基-丁二酸-1,4 单酯)-4-]-β-环糊精键合全多孔硅胶固定相分别在正相和反相模式下进行了手性药物扑尔敏的分离研究.建立了手性药物扑尔敏拆分的高效液相色谱检测方法.在正相最佳条件下分离度Rs为1.37;反相最佳条件下分离度Rs达10.58.考察了流动相配比、pH值对分离效果的影响.确定在反相模式下扑尔敏浓度为0.000~1.367 mg/mL范围内,峰高、峰面积与浓度呈线性关系.通过精密度试验考察,相对标准偏差RSD(n=12)均小于4%.加标回收率为86.14% ~108.76%.加标浓度为0.20 mg/mL时,以峰高-浓度曲线计算,回收率为94.69% ~106.93%.
[1]Chen Q C,Jeong S J,Hwang G S,et al.Enantiose lective determination of chlorpheniramine in various formulations by HPLC using carboxymethyl-β-cyclodextrin as a chiral additive[J].Archives of Pharmacal Research,2008,31(4):523-529.
[2]Haginaka J,Kagawa C.Uniformly sized molecularly imprinted polymer for d-chlorpheniramine evaluation of retention and molecular recognition properties in an aqueousmobile phase[J].Journal of Chromatography A,2002,948:77-84.
[3]Fried K M,Young A E,Yasuda S U,et al.The enantioselective determination of chlorpheniramine and its major metabolites in human plasma using chiral chromatography on aβ-cyclodextrin chiral stationary phase and mass spectrometric detection[J].Journal of Pharmaceutical and Biomedical Analysis,2002,27:479-488.
[4]Wang Huaisong,Xu Dan,Jiang Ping,et al.Novel restricted access chiral stationary phase synthesized via atom transfer radical polymerization for the analysis of chiral drugs in biologicalmatrices[J].Analyst,2010,135:1785-1792.
[5]杨 曦,沈静茹,李 云.双[-6-氧-(-3-间硝基苯磺酰基-丁二酸-1,4 单酯)-4-]-β-环糊精的合成及模拟抗坏血酸氧化酶[J].中南民族大学学报:自然科学版,2006,25(4):28-30.
[6]Wind M,Hoffmann P,Wagner H,etal.Chiral capillary electrophoresis as predictor for separation of drug enantiomers in continuous flow zone electrophoresis[J].Journal of Chromatography A,2000,895:51-65.
