一种蛋白质质谱分析中快速胍基化新方法
2011-02-02王雪颖张养军
王雪颖,张养军,王 昕,3,
佟 巍2,秦伟捷2,毛心丽1,2,钱小红2
(1.沈阳药科大学,辽宁 沈阳 110015;2.军事医学科学院放射与辐射医学研究所,北京蛋白质组研究中心,蛋白质组学国家重点实验室,北京 102206;3.防化指挥工程学院,北京 102205)
一种蛋白质质谱分析中快速胍基化新方法
王雪颖1,2,张养军2,王 昕2,3,
佟 巍2,秦伟捷2,毛心丽1,2,钱小红2
(1.沈阳药科大学,辽宁 沈阳 110015;2.军事医学科学院放射与辐射医学研究所,北京蛋白质组研究中心,蛋白质组学国家重点实验室,北京 102206;3.防化指挥工程学院,北京 102205)
胍基化修饰在蛋白质组学鉴定及定量研究中发挥着重要的作用,本研究建立了一种蛋白质质谱分析中微波辅助快速胍基化的新方法。采用标准肽段系统地优化了反应溶剂p H值、O-甲基异脲的浓度及微波加热时间,发现标准肽段在p H 12的氨水溶液中,O-甲基异脲的浓度为1.5 mol/L,微波高火加热1 min的胍基化效率为99%。将此方法应用于马心肌红蛋白和牛血清白蛋白胰蛋白酶酶切肽段,质谱检测结果表明,含赖氨酸肽段的平均胍基化效率(95%)大于文献报道的平均胍基化效率(85%)。与传统水浴加热胍基化方法相比,微波辅助加热胍基化方法缩短了反应时间,提高了反应效率,降低了肽段N端副反应。
胍基化;微波辅助;肽;蛋白质定量
胍基化是多肽和蛋白质重要的化学修饰之一。经胍基化反应,多肽的赖氨酸(lysine)转化为高精氨酸,其原理示于图1。胍基化反应被广泛应用于提高多肽在基质辅助激光解吸电离(MALDI)质谱中的离子化效率和信号强度[1-2];改变多肽在质谱中的碎片裂解效率以改善多肽的二级谱图质量和提高多肽的覆盖率[3];或根据研究的需要特异性地封闭赖氨酸的ε-氨基[4-6]。胍基化反应在蛋白质组学的相对定量研究中也有应用,例如Carabetta等[7]采用金属内切蛋白酶Lys-N酶切方法结合15N/14N标记的O-甲基异脲试剂进行胍基化反应,在提高了离子化效率和检测灵敏度的同时,实现了对大肠杆菌(E.coli)中多A聚合酶I(PAPI)蛋白质的相对定量。但是,目前报道的各种胍基化方法仍存在不足,一是胍基化反应时间长,文献报道的有30 min[8]、2 h[5-6]、过夜[7]甚至4天的反应时间[9];二是胍基化的反应效率还有待提高,Beardsley等[10]考察了血红蛋白(hemoglobin)、牛血清白蛋白(bovine serum albumin)和新月柄杆菌(caulobacter crescentus)中的ctr A蛋白酶切肽段的胍基化反应情况,3种蛋白酶切肽段的平均胍基化效率仅为85%。
为了解决这些问题,本研究选用具有挥发性的氨水为胍基化碱性溶剂,通过微波高火加热的方式,建立一种新型快速的胍基化方法,并与传统水浴加热方式做比较。
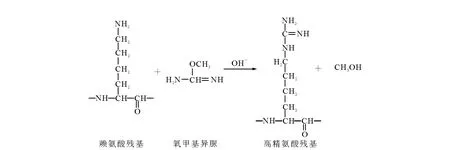
图1 胍基化反应原理示意图Fig.1 The reaction schematic diagram of guanidination
1 实验部分
1.1 仪器与装置
4800基质辅助激光解吸电离飞行时间串联质谱仪:美国 ABsciex公司产品;Sartorius BP211d分析天平:瑞士Sartorius公司产品;Thermo Orion MODEL 818 p H计:美国Thermo公司产品;微波炉:广东Galanz公司产品;C18 zip tip脱盐柱:美国Promega公司产品。
1.2 材料与试剂
标准肽段(色谱方法测定纯度为99%,绝对含量70%):上海吉尔生化公司产品;马心肌红蛋白、牛血清白蛋白:美国Sigma公司产品;α-氰基-4-羟基肉桂酸(St.Louis,MO):美国Sigma公司产品;二硫苏糖醇和测序级胰蛋白酶(Madison,WI):美国Promega公司产品;三氟乙酸、O-甲基异脲半硫酸盐、碘乙酰胺:美国 Acros Organics公司产品;乙腈(色谱纯,Phillipsburg,ΜSA):美国J.T.Baker公司产品;乙酸、氨水等其他试剂为分析纯:北京化学试剂公司产品。实验用水经 Milli-Q A10型纯水仪制备(Bedford,MA):美国Millipore公司产品。
1.3 标准肽段的胍基化反应条件优化
取1μL 1 g/L的标准肽段(DVDPGEHYIIK,相对分子质量1 285.64),加入5μL 25%的氨水,依次改变O-甲基异脲终浓度(50,100,200,400,600,1 000,1 500 mmol/L)、溶液 p H值(9.0,10.0,10.5,11.0,11.5,12.0,13.0)和微波高火加热时间(15,30,60,90,120 s)等反应条件,再加入25μL 10%三氟乙酸(TFA)中止反应,然后进行质谱分析,计算峰强度比值H1327/(H1327+H1285),来评估标准肽段的胍基化反应程度及特异性。
1.4 蛋白质酶切
将100μg马心肌红蛋白(Myoglobin)溶于100μL 50 mmol/L碳酸氢铵缓冲液中,95℃加热变性10 min后,冷却到室温,加入2μg胰蛋白酶,37 ℃ 孵育,分别在 5、10、15、20、25、30 min,1、2 h,过夜等不同时间段后各取5μL酶切液,然后加入100 m L水,微波高火加热到95℃,加入酶切样品,低火微波加热5~10 min,中止酶切反应。最后将各时间点的酶切液混合。
将100μg牛血清白蛋白(BSA)溶解于50 μL 8 mol/L 尿 素 (含 10 mmol/L DTT,50 mmol/L NH4HCO3缓冲液,p H 8.0)中,37 ℃温育1 h,加入IAA使其终浓度为50 mmol/L,室温放置于暗处2 h,加入50 mmol/L缓冲液使反应体系中尿素终浓度小于1 mol/L,加入2μg胰蛋白酶,37℃孵育过夜,微波辅助加热中止酶切反应。
1.5 传统水浴胍基化与微波辅助加热胍基化
1.5.1 传统水浴加热胍基化方法[11]取1μL 1 g/L马心肌红蛋白酶切液,加入5.5μL 7 N氨水混匀,再加入1.5μL 7.9 mol/LO-甲基异脲,涡旋混匀,65℃水浴恒温5~10 min,最后加入15μL 10%的TFA终止反应。用Zip Tip C18反相小柱脱盐,分别用100%和50%乙腈冲洗小柱,然后用10μL 0.1%TFA平衡脱盐柱,上样10μL,最后用5μL 80%ACN/0.1%TFA 洗脱。脱盐完成后进行质谱检测。
1.5.2 微波辅助加热胍基化方法 取1μL 1 g/L马心肌红蛋白酶切液,加入5μL 25%的氨水(p H 12),涡旋混匀,加入新鲜配制的O-甲基异脲,使其终浓度为1.5 mol/L,混匀后高火微波1 min。然后加入25μL 10%的TFA终止反应。用Zip Tip C18反相小柱脱盐,进行质谱检测。
1.6 牛血清白蛋白胰蛋白酶酶切肽段胍基化反应
取上述1μL 1 g/L牛血清白蛋白胰酶酶切肽段,加入5μL p H 12的氨水,涡旋混匀,再加入现配制的O-甲基异脲,使其终浓度为1.5 mol/L,混匀后高火微波1 min。然后加入25μL 10%的TFA终止反应。用Zip Tip C18反相小柱脱盐,取反应产物进行质谱分析,计算峰强度比值H原肽段/(H原肽段+H胍基化肽段),来评估肽段的胍基化反应效率及特异性。
1.7 质谱分析条件
待测样品混合基质(α-氰基-4-羟基肉桂酸)溶液点靶分析。上样量为1 pmol,马心肌红蛋白胰酶酶切肽段作为标准物对仪器进行外标校正,校准至误差≤0.1 u,相对标准偏差≤10 ppm。一级质谱数据采集使用MS-positive反射模式,扫描范围600~3 500,激光能量5 000,每张谱图累加800次。二级质谱数据采用MS/MS-1 k V反射模式,从一级谱图中选择信噪比大于20的峰,母离子选择范围设定其相对分辨率为150,激光能量为5 500,每张谱图累加1 200次。仪器控制软件为4800Exp LorerTMsoftware。本研究所有质荷比(m/z)均为单同位素峰质量数。
2 结果与讨论
2.1 标准肽段胍基化反应条件优化
影响胍基化反应的主要因素包括:反应体系的p H值、O-甲基异脲的浓度、反应的温度等。本研究通过改变传统的水浴加热方式,采用高火微波加热方式,并从溶剂种类、反应溶剂p H值、O-甲基异脲的浓度、微波时间等方面对胍基化反应进行了优化。
2.1.1 胍基化反应溶剂的选择 胍基化反应在碱性环境中进行,常用的碱性试剂有氢氧化钠[2,12]、碳酸钠[1,3]、氨水[11,13]。由于氨水具有挥发性,容易从反应体系中除去,可减少质谱分析时[M+Na]+峰的干扰,故本实验选用氨水为反应溶剂。
2.1.2 反应溶液p H的优化 反应体系的p H值是影响胍基化效率的关键因素之一。从反应原理分析,在强碱性条件下有利于赖氨酸ε-氨基(p Ka=10.53)的去质子化从而使反应顺利进行;同时鉴于在p H大于13.6时多肽将发生水解副反应[12],实验考察了在25%的氨水溶剂中,p H值在9~13.5,O-甲基异脲的终浓度为1.5 mol/L,微波辅助加热1 min条件下的胍基化反应效率,反应结果示于图2a。随着反应体系的p H增加至12,胍基化的反应效率达到最大值97%,p H值再增加则胍基化效率几乎保持不变。所以,最佳的p H值是11.5~13,此区间标准肽段的胍基化反应效率可达95%以上。
2.1.3O-甲基异脲浓度的优化 文献报道常用的胍基化试剂有:O-甲基异脲硫酸氢盐、O-甲基异脲半硫酸盐、氨基氰、吡唑-1-羧酰胺、S-甲基-硫脲及其衍生物[14],因O-甲基异脲硫酸氢盐或O-甲基异脲半硫酸盐对赖氨酸侧链反应的专一性强,故多选择O-甲基异脲试剂。另外O-甲基异脲硫酸氢盐是由一分子HSO4-和一分子O-甲基异脲组成,其饱和溶液p H值为0.76;而O-甲基异脲半硫酸盐是由一分子O-甲基异脲和1/2分子SO42-组成,其饱和溶液p H 值为2.60[11]。O-甲基异脲半硫酸盐对胍基化反应体系的p H值影响小,故选用此试剂。
O-甲基异脲半硫酸盐的浓度也是直接影响胍基化效率的重要因素之一。图2b显示了25%氨水溶液、p H值为12时,O-甲基异脲半硫酸盐的终浓度从0.1~1.5 mol/L变化所对应的胍基化效率,在1 mol/L和1.5 mol/L时,胍基化效率为96%以上,终浓度为1.5 mol/L时,反应效率最高为99%。
2.1.4 微波辅助加热时间的优化 缩短化学反应的时间以减少由于反应时间长而引起的副反应、提高研究的效率和加快整个研究的进程等为优化反应条件的共同目标之一。传统的缩短胍基化反应时间的手段是水浴加热,如Chervenka等[9]对胰凝乳蛋白酶原(chymotrypsinogen)进行胍基化反应时,在4℃反应4天;当反应温度提高到37℃时,反应时间缩短到2 h[5]。Rich-ard等[11]在水浴65℃时对肽段进行胍基化,反应时间缩短到5~10 min。
微波加热不同于一般的温度从高向低的热传导过程,温度的升高直接发生于被加热物体内部而不是发生于加热腔内或是在容器上,所以微波加热具有加热速度快,加热均匀的特点。本实验主要考察了标准肽段微波高火15、30、60、90、120 s的反应,结果示于图2c,发现标准肽段在30 s~2 min胍基化效率都达96%以上,反应时间低于文献报道的5~10 min[11]。分析原因,可能是微波加热提高了反应速率。
2.2 水浴加热与微波辅助加热胍基化方法的比较
选用肌红蛋白胰酶酶切肽段为模型,按文献[11]报道的水浴方法与微波辅助胍基化方法分别进行胍基化反应。并按不同时间点将酶切所产生的含漏切位点的马心肌红蛋白肽段与过夜酶切的肽段混合,以便考察微波辅助胍基化方法对含漏切位点肽段和无漏切位点肽段胍基化的反应效率。
在25%氨水(p H 12),O-甲基异脲终浓度为1.5 mol/L,微波高火加热1 min的条件下反应,马心肌红蛋白酶切肽段胍基化反应结果示于表1和图3,水浴方法与微波辅助胍基化方法都能使无漏切位点肽段(peptide 1-7)胍基化效率达到98%以上,对含有漏切位点的肽段(peptide 8-14)胍基化效率可达到95%,但微波辅助加热的方法更占优势。随着赖氨酸数量的增多,肽段的(peptide 16)胍基化反应效率降低。所选肽段胍基化反应的标准偏差小于15%。

图2 不同条件对标准肽段胍基化反应效率的影响a.氨水的p H值;b.O-甲基异脲的浓度;c.微波加热时间Fig.2 Different reaction conditions on guanidination efficiencies of standard peptide a.p H of ammonia;b.concentration of O-methylisourea hemisulfate;c.time of microwave heating
在对比实验中,发现N端伯胺基也会发生修饰反应。N端伯胺基与O-甲基异脲的反应与N端氨基酸种类有关,在马心肌红蛋白酶切肽段微波加热方法中,以Y、L、H、F、A、G氨基酸为N端的肽段都发生了副反应。其中以A(peptide 1)、G(peptide 6)为 N 端的肽段的胍基化效率分别为5%和16%。Y、L、H、F氨基酸为N端的氨基胍基化效率在1.5%以下,可忽略。A和G为N端的肽段胍基化的原因可能是结构简单,空间位阻小,导致副反应的可能性增大。而这两个肽段在水浴加热方式反应中副反应分别为7%和22%。据文献报道[11],在马心肌红蛋白和细胞色素C酶切肽段水浴加热方式胍基化数小时,M、S、V、L、F、E、A 都会发生副反应。相比之下,由于微波辅助胍基化可在1 min完成,因此副反应更少。
在分析肽段N端副反应时,其中以A为N端的肽段(peptide 1)质量数为748.4,N端被胍基化后质量数增加为790.4,同时肌红蛋白含一个漏切位点的肽段质量数增加也为790.4,为了验证748.4的N端是否被胍基化,在 MALDITOF-TOF质谱上做了二级质谱分析,比较b、y离子是否匹配。当N端被修饰,b离子质荷比增加42,而y离子不变。y离子的信号强于b离子,所以实验着重观察了y离子是否匹配。如图4所示,实验发现790.4肽段的y离子与748.4完全匹配,从而证明了以丙氨酸为N端的肽段的确发生了胍基化反应。其他N端是否发生副反应也可以通过此方法进行验证。

图3 马心肌红蛋白胰酶酶切肽段胍基化前(a)和胍基化后(b)的 MALDI-TOF/TOF-MS图Fig.3 MALDI-TOF/TOF mass spectra of peptides from myoglobin before guanidination(a)and after microwave-assisted guanidination(b)
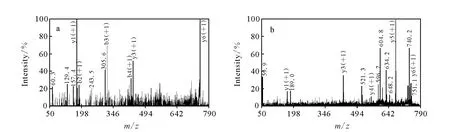
图4 马心肌红蛋白酶切肽段T135-140(ALELFR)N端被修饰前(a)和被修饰后(b)的 MALDI-TOF/TOF-MS的二级图Fig.4 MALDI-TOF/TOF MS/MS spectra of pepetide1 before(a)and after(b)guanidination


2.3 微波辅助加热胍基化方法的验证
为了进一步验证微波辅助加热胍基化方法的可靠性,选择牛血清白蛋白(BSA)胰蛋白酶酶切肽段进行胍基化反应,实验结果列于表2。在质谱所鉴定到的26个以赖氨酸为C端的肽段中,其中有11个肽段的胍基化效率达98%以上,8个肽段的胍基化达到90%以上,6个肽段的胍基化效率82%以上,1个含脯氨酸肽段的胍基化为50%。所选肽段胍基化反应的标准偏差均小于20%。证明了本实验建立的胍基化方法的普适性。实验所选择的两个蛋白质(马心肌红蛋白和牛血清白蛋白)含赖氨酸肽段的平均胍基化效率(95%)大于文献报道的平均胍基化效率(85%)[10]。 另 外, 肽 段 (RPCFSALTPDETYVPK)的胍基化效率仅为50%,其原因可能是脯氨酸的吡咯环的空间位阻大,影响赖氨酸侧链氨基的胍基化反应。
对于肽段N端副反应的考察,不仅观察了BSA以R结尾的肽段,也考察了理论酶切肽段赖氨酸侧链氨基完全胍基化后的N端是否发生修饰。实验发现此副反应不但与N端氨基酸的种类有关,也与肽段本身的结构有关。其中以L、E、G、S氨基酸为N端的肽段,副反应胍基化效率超过15%,以A为N端的肽段副反应为5%。除了以 W、P、I、F这4个氨基酸为N端的肽段在BSA的肽段没有考察到,其余以D、H、M、V、T、C、N、R、Q、Y为 N 端的肽段没有发现胍基化副反应。
3 结论
本实验改进了传统胍基化的方法。采用微波辅助加热1 min,在p H 11.5~13的氨水溶液中,O-甲基异脲半硫酸盐的终浓度在1 mol/L和1.5 mol/L时,马心肌红蛋白和牛血清白蛋白胰酶酶切且含赖氨酸肽段的平均胍基化效率(95%)大于文献报道的平均胍基化效率(85%)。与水浴加热胍基化方法相比,微波辅助加热胍基化方法缩短了反应时间,提高了反应效率,降低了肽段N端副反应,使得本方法在蛋白质定性定量分析中发挥重要的作用。
[1]HALE J E,BUTLER J P,KNIERMAN M D,et al.Increased sensitivity of tryptic peptide detection by MALDI-TOF mass spectrometry is achieved by conversion of lysine to Homoarginine[J].Anal Biochem,2000,287(1):110-117.
[2]BRANCIA F L,OLIVER S G,GASKELL S J.Improved matrix-assisted laser desorption/ionization mass spectrometric analysis of tryptichydrolysates of proteins following guanidination of lysine-containing peptides[J].Rapid Commun Mass Spectrom,2000,14(21):2 070-2 073.
[3]HENNRICH M L,BOERSEMA P J,VAN DEN TOORN H,et al.Effect of chemical modifications on peptide fragmentation behavior upon electron transfer induced dissociation[J].Anal Chem,2009,81(18):7 814-7 822.
[4]林虹君,卫军营,贾 伟,等.胍基化修饰结合稀土金属标记应用于蛋白质相对定量研究[J].质谱学报,2010,31(5):284-290.
[5]ZHOU C,ZHANG Y,QIN P,et al.A method for rapidly confirming protein N-terminal sequences by matrix-assisted laser desorption/ionization mass spectrometry[J].Rapid Commun Mass Spectrom,2006,20(19):2 878-2 884.
[6]ZHAO L,ZHANG Y,WEI J,et al.A rapid isolation and identification method for blocked N-terminal peptides by isothiocyanate-coupled magnetic nanoparticles and MS[J].Proteomics,2009,9(18):4 416-4 420.
[7]CARABETTA V J,LI T,SHAKYA A,et al.Integrating Lys-N proteolysis and N-terminal guanidination for improved fragmentation and relative quantification of singly-charged ions[J].J Am Soc Mass Spectrom,2010,21(6):1 050-1 060.
[8]WANG DXU W,MCGRATH SC,PATTERSON C,et al.Direct identification of ubiquitination sites on ubiquitin-conjugated CHIP using MALDI mass spectrometry[J].J Proteome Res,2005,4(5):1 554-1 560.
[9]CHERVENKA C H,WILCOX P E.Chemical derivatives of chymotrypsinogenⅡreaction withO-methylisourea [J].J Biol Chem,1956,222:635-647.
[10]BEARDSLEY R L,KARTY J A,REILLY J P.Enhancing the intensities of lysine-terminated tryptic peptide ions in matrix-assisted laser desorption/ionization mass spectrometry[J].Rapid Commun Mass Spectrom,2000,14(23):2 147-2 153.
[11]BEARDSLEY R L,REILLY J P.Optimization of guanidination procedures for MALDI mass mapping[J].Anal Chem,2002,74(8):1 884-1 890.
[12]KEOUGH T,LACEY M P,YOUNGQUIST R S.Derivatization procedures to facilitate de novo sequencing of lysine-terminated tryptic peptides using postsource decay matrix-assisted laser desorption/ionization mass spectrometry[J].Rapid Commun Mass Spectrom,2000,14(24):2 348-2 356.
[13]COCKRILL S L,FOSTER K L,WILDSMITH J,et al.Efficient micro-recovery and guanidination of peptides directly from MALDI target spots[J].Biotechniques,2005,38(2):301-304.
[14]RODRIGUEZ F,ROZAS I,ORTEGA J E,et al.Guanidine and 2-Aminoimidazoline Aromatic Derivativesα2-adrenoceptor antagonists,1:toward new antidepressants with heteroatomic linkers[J].J Med Chem,2007,50(18):4 516-4 527.
A Novel Method of Fast Guanidination in Protein Analysis
WANG Xue-ying1,2,ZHANG Yang-jun2,WANG Xin2,3,TONG Wei2,QIN Wei-jie2,MAO Xin-li1,2,QIAN Xiao-hong2
(1.Shenyang Pharmaceutical University,Shenyang 110015,China;2.State Key Laboratory of Proteomics,Beijing Proteome Research Center,Beijing Institute of Radiation Medicine,Beijing 102206,China;3.Institute of Chemical Defense,Beijing 102205,China)
Guanidination plays an obvious role in identification and quantification of proteins.A quick microwave-assisted guanidination reaction method was established in this work.The reaction conditions,including buffer p H,concentration ofO-methylisourea hemisulfate and time of microwave heating,were optimized using a standard peptide.The peptide was microwave heated for 1 min with 1.5 mol/LO-methylisourea and near p H 12 of the mixture,the conversion rate of lysine to homoarginine was 99%.Further examination was carried out by applying this microwave-assisted guanidination method to tryptic digested peptides of myoglobin and bovine albumin.The average guanidination efficiency washigher than that reported in literature.Compared with the conventional guanidination methods,the microwave-assisted guanidination method can shorten reaction time,enhance reaction efficiency,and reduce side reactions on the N-terminal of peptides.
guanidination;microwave-assisted;peptides;protein quantification
O 657.63
A
1004-2997(2011)05-0257-08
2011-02-14;
2011-06-01
国家高技术研究发展计划(2006AA02A308)、国家自然科学基金项目(30621063,20635010,20735005,20875101)、973计划(2007CB914104、2010CB912701、2011CB910603)资助
王雪颖(1988~),女(汉族),天津人,硕士研究生,药物分析专业。E-mail:piaopiaoxiaoxue@126.com
钱小红(1955~),女(汉族),江苏人,研究员,从事蛋白质组学研究。E mail:qianxh1@yahoo.com.cn
