利用分枝杆菌的重组工程系统将TAP标签敲入耻垢分枝杆菌的方法研究*
2011-01-24赵丽丽赵秀芹万康林
赵丽丽,夏 强,2,赵秀芹,万康林
利用分枝杆菌的重组工程系统将TAP标签敲入耻垢分枝杆菌的方法研究*
赵丽丽1,夏 强1,2,赵秀芹1,万康林1
目的 利用pJV 53质粒编码的分枝杆菌重组工程系统将TAP标签敲入耻垢分枝杆菌基因组。方法将pJV 53质粒转入耻垢分枝杆菌,使其表达重组蛋白,制备带有重组蛋白的感受态细胞;从质粒pBS1479中扩增出 tap片段,从质粒pSM T3中扩增hyg片段,从耻垢分枝杆菌基因组中分别扩增出aasf基因及其5′端非编码区片段AASF5和aasf基因下游3′端非编码区片段AASF3,利用重叠PCR将以上4个片段拼接在一起,形成最终的敲入片段A 5THA 3;将构建好的敲入片段转入感受态细胞,使其重组入耻垢分枝杆菌基因组中,PCR和DNA测序鉴定敲入效果。结果重叠 PCR技术得到全长为3 200 bp的敲入片段,PCR与DNA测序结果证实带有TAP标签的A 5THA 3片段已成功敲入耻垢分枝杆菌基因组中。结论成功将 TAP标签敲入耻垢分枝杆菌基因组,为下一步进行目的基因功能研究奠定了基础。
重组工程系统;pJV 53质粒;串联亲和纯化标签;敲入片段;重叠PCR;耻垢分枝杆菌
“重组工程”是近几年兴起的一种新型体内同源重组的遗传工程技术,它是由噬菌体重组酶介导的体内同源重组,可有效促进线性DNA分子和染色体间的重组,能直接在体内对染色体DNA进行基因敲除、敲入或替换。
pJV 53质粒[1]是一种大肠杆菌-分枝杆菌穿梭载体,能编码产生Che9c分枝杆菌噬菌体的gp60和gp61蛋白,它们分别是重组蛋白 RecE和 Rec T的同源物,能帮助外来基因序列重组入分枝杆菌,且所需同源序列短,重组效率高;aasf基因编码产生AASF蛋白,它在耻垢分枝杆菌中的功能尚不清楚;TAP-tag的敲入是进行串联亲和纯化技术(tandem affinity purification,TAP)[2-3]的关键,TAP技术是近年来出现的一种能快速研究生理条件下蛋白质相互作用的新方法,该技术有助于我们进一步认识蛋白质的功能。
本研究首先将pJV 53质粒转入耻垢分枝杆菌M.smegm atism c2155,加入诱导剂,使其表达重组工程系统——gp60和gp61,制备带有 gp60和gp61的耻垢分枝杆菌感受态细胞;利用overlap PCR技术将tap序列、hyg序列(用作选择性标记)和目的基因(aasf)及其3′端非编码序列(用作重组)拼接在一起形成最终的敲入(knock-in)片段A 5THA 3(见图1);将A 5THA 3转入感受态细胞,在重组蛋白gp60和gp61的帮助下,A 5THA 3成功敲入待研究的耻垢分枝杆菌靶基因(aasf)处,为下一步利用TAP技术研究靶基因的功能奠定基础。

图1 Overlap PCR构建TAP敲入片段Fig.1 Construction of tap TAP knock-in cassette by overlap PCR
1 材料与方法
1.1 材料
1.1.1 质粒与菌株pBS1479 质粒(含 tap基因)购于 EUROSCARF,pSM T3质粒(含 H ygrom ycin B抗性基因,hyg)由伦敦帝国理工学院Robertson博士惠赠,大肠杆菌-分枝杆菌穿梭质粒pJV 53由匹兹堡大学的 Hatfull教授惠赠,M.smegm atis m c2155购自A TCC,DH5a大肠杆菌为本室保存。
1.1.2 主要试剂 Pyrobest DNA聚合酶,Bam H I,H in d III,T4DNA连接酶购自 Takara公司,质粒小提试剂盒、PCR产物回收试剂盒、胶回收试剂盒、Tarns2KTMPlusⅡDNA M arker购自北京全式金生物技术有限公司。
1.2 方法
1.2.1 制备重组工程菌株将质粒 pJV 53通过电击转化转入M.smegm atism c2155感受态细胞中,电击参数为:电压 2.5 kV,电阻1000Ω,电容 25 μF。电击过后,立即加入1 m L的7H9培养基,37℃振荡培养2 h,立即涂于7H10(含 KAN,OADC)抗性平板[4]。
1.2.2 制备重组工程菌株感受态细胞 挑取新鲜的含有pJV 53质粒的单菌落接种于5 mL 7H9培养基(含 KAN,ADC和0.05%Tween-80)中,37 ℃振荡培养至OD600值0.6左右,接种至200 m L的7H9培养基(成分同上),37℃过夜培养至OD600值0.4左右,加入乙酰胺(0.2%),继续培养2~3 h。将培养物冰上放置0.5~1 h,4℃,5 000 r/min离心10 min收集菌体,将菌体用100 m L预冷的10%的无菌甘油重悬,4℃,5 000 r/m in离心10 m in,收集菌体,重复清洗菌体3次,所用10%的甘油体积依次减为50 m L,25 m L,12.5 m L,最后加入5 m L预冷的10%的甘油,吹匀菌体后,以0.2 mL/管分装保存于-80℃。
1.2.3 利用overlap PCR技术扩增敲入片段 反应分几步进行:
①从质粒pBS1479中扩增出 tap片段(作为②的上游片段),长度约为600 bp,引物为 Tap-5和Tap-3;从质粒p SM T3中扩增 hyg片段(作为②的下游片段),长度约为 1 200 bp,引物为 Hyg-5和Hyg-3;从耻垢分枝杆菌基因组中分别扩增出 aasf基因及部分5′端非编码区片段AASF5,长度约为650 bp,引物为AASF5-5和AASF5-3,aasf基因下游3′端非编码区片段AASF3,长度约为550 bp,引物为AASF3-5和AASF3-3,PCR反应体系总体积均为50μL,其中 10×Pyrobest PCR Buffer 5μL,dN TP(2.5 mmol)4μL,Forward Primer(20 mmol)1μL,Reverse Primer(20 mmol)1μL,DNA模板 1μL,Pyrobest DNA Polymerase 0.3 μL,无菌双蒸水37.7μL,PCR反应条件均为:95℃5 min,94℃1 min,64℃1min,72℃1 min 30 s,25次循环;72℃5min。
②通过overlap PCR将 tap基因和抗性基因hyg连接起来,形成片段 TH,长度约为1 800 bp,所用引物为 Tap-5和 Hyg-3。反应体系总体积均为50μL,其中 10×Pyrobest PCR Buffer 5μL,dN TP(2.5 mmol/L)4μL,Forward Primer(20 mmol/L)1μL,Reverse Primer(20 mmol/L)1μL,上游片段 1μL,下游片段 1μL,Pyrobest DNA Polymerase 0.3μL,无菌双蒸水36.7μL。PCR反应条件为:95℃5 min;94℃1 min,64℃1 min,72℃2 min,25次循环;72℃5 min。
③利用同样方法,将 TH片段与AASF3相连接,形成片段 THA 3。上游片段引物为 Tap-5和H3,模板为 TH片段,PCR反应体系同①,PCR反应条件同②。下游片段引物为 H 5和AASF3-3,模板为AASF3片段,PCR反应体系和条件均同①。THA 3片段引物为 Tap-5和AASF3-3,PCR反应体系同②。PCR反应条件为:95℃5 min;94℃1 min,64℃1 min,72℃3 min,25次循环;72℃5 min。
④扩增全长敲入片段(A 5THA 3)。上游片段引物为 AASF5-5和 AASF-U 3,模板为 AASF5片段,PCR反应体系和条件均同①,下游片段引物为AASF-D5和 AASF3-3,模板为 THA 3片段,PCR反应体系同 ①,PCR反应条件同 ②。扩增A 5THA 3片段的PCR反应体系同②,PCR反应条件为:95℃5 min;94℃1 min,62℃1min,72℃4m in,25次循环,72℃5 min。本研究所涉及的引物序列见表1。

表1 引物序列Table 1 The primer sequence
1.2.4 通过重组系统将构建好的A 5THA 3片段敲入基因组中的相应位置 将A 5THA 3片段进行胶回收纯化,吸取100 ng纯化产物电击转化入重组工程菌株感受态细胞中,37℃振荡培养4 h,然后将转化产物涂平板(含有 Kan+Hyg抗性),37℃倒置培养3~5 d后,挑取阳性克隆进行菌体PCR验证。1.2.5 PCR和基因测序验证重组子 分别用引物AASF5-5,Tap-3和AASF5-5,AASF-D5进行菌液PCR扩增,反应体系为 50μL,其中 10×Pyrobest PCR Buffer 5μL,dN TP(2.5 mmol/L)4μL,Forw ard Primer(20 mmol/L)1μL,Reverse Primer(20 mmol/L)1μL,菌液 1μL,Pyrobest DNA Polymerase 0.3μL,无菌双蒸水 37.7μL。PCR反应条件为:95℃5 min;94℃1 min,64℃1 min,72℃3 min,30次循环;72℃5 min。将PCR产物纯化并送擎科生物技术公司测序。
2 结 果
2.1 利用overlap PCR技术得到 TH片段 分别以PCR扩增出的 tap和 hyg为模板,Tap-5和Hyg-3为引物,overlap PCR得到 TH片段,长度大约为1 800 bp,如图2。
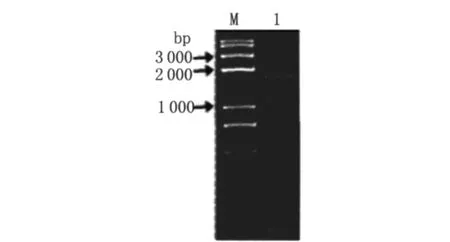
图2 overlap PCR得到TH片段Fig.2 Overlap PCR product of TH fragment M:DNA marker 1:TH fragment
2.2 利用overlap PCR技术得到 THA 3片段 以TH片段为模板,Tap-5和 H3为引物,PCR得到上游片段;AASF3片段为模板,H5和AASF3-3为引物,PCR得到下游片段;以上下游片段为模板,Tap-5和AASF3-3为引物,PCR得到 THA 3片段,长度大约为2 400 bp,如图3。
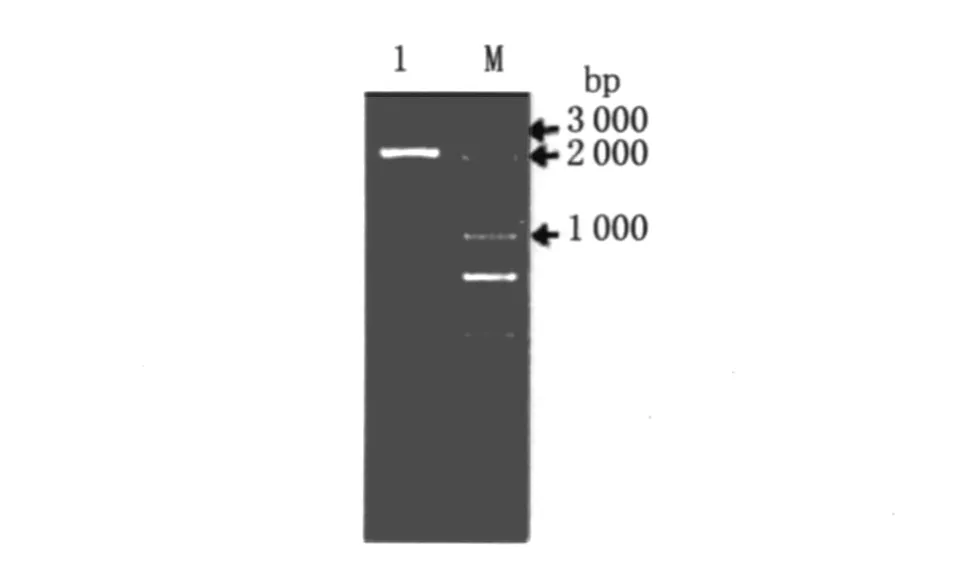
图3 overlap PCR得到THA3片段Fig.3 Overlap PCR product of THA3 fragment M:DNA marker 1:THA 3 fragment
2.3 利用overlap PCR技术得到A 5THA 3片段以AASF5片段为模板,AASF5-5和AASF-U 3为引物,PCR得到上游片段;THA 3片段为模板,AASF-D5和AASF3-3为引物PCR得到下游片段;以上下游片段为模板,AASF5-5和AASF3-3为引物,PCR得到 THA 3片段,长度大约为3 100 bp,如图4。
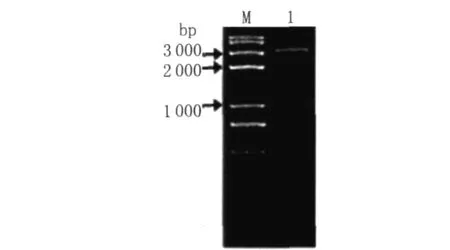
图4 overlap PCR得到敲入片段A5THA3Fig.4 Overlap PCR product of knock-in fragmen t A5THA3 M:DNA marker 1:A 5THA 3 fragment
2.4 将A 5THA 3片段敲入耻垢分枝杆菌的 aasf基因位置 将A 5THA 3敲入片段胶回收纯化,电击转化入耻垢分枝杆菌细胞内,利用新发展起来的基于分枝杆菌的重组工程系统,将A 5THA 3片段整合到耻垢分枝杆菌基因组的aasf基因处,利用敲入片段上的潮霉素抗性基因筛选阳性克隆,再对筛选出的克隆进行PCR检测以验证重组子是否发生重组。其中的一条引物是AASF5-5,另一条引物是Tap-3,只有整合了 TAP-tag的重组子才能扩增出相应大小的条带,而野生型菌是无法扩增出条带的,如图5。
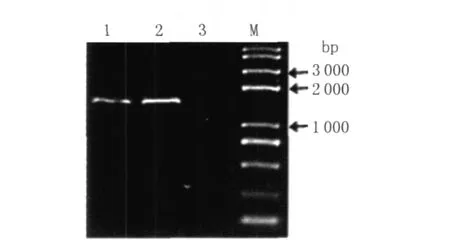
图5 PCR验证重组子Fig.5 Identification of the recombinants by PCR M:DNA marker;1,2:recombinants;3:wild-type
同时,利用两条外侧引物AASF5-5和AASFD5进行PCR扩增时,重组子由于敲入了带有 TAP标签和抗性基因的 TH片段,与野生型相比,就多出这段外源序列,图6验证了这一结果,证明已成功将TAP-tag片段敲入耻垢基因组中 aasf基因的C端,测序结果也显示tap片段成功整合在aasf基因的 3′端。
3 讨 论
串联亲和纯化技术(tandem affinity purification,TAP),特别适用于研究蛋白质在生理条件下的相互作用。该技术的关键是将一个纯化标签(TAP-tag)引入靶蛋白质一端,不破坏靶蛋白质的序列,且不改变靶蛋白在生物体内的表达水平,经过亲和纯化获得接近自然条件的特定蛋白质复合体,以便于下一步的分离鉴定,目前,TAP技术已成功运用于大肠杆菌、酵母、昆虫细胞及哺乳动物细胞,但在分枝杆菌菌体内,还未有这方面的应用报道。
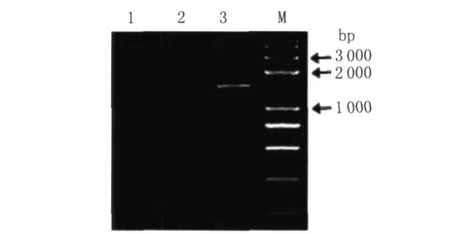
图6 PCR验证重组子Fig.6 Identification of the recombinants by PCR M:DNA marker;1,2:recombinants;3:wild-type
重组工程系统最早应用于大肠杆菌,包括基于缺陷型λ噬菌体的Red重组系统[5-6]和依赖Rac噬菌体的ET重组系统[7]。其中,Rac噬菌体的ET重组系统编码基因为 rec E和rec T,编码产生的RecE蛋白具有核酸外切酶活性,能从5′端向3′端降解双链DNA分子,产生3′突出端;Rec T蛋白能结合在单链DNA 3′突出端,防止其被单链核酸酶降解,同时介导互补单链DNA的退火。整个重组过程无需使用限制性内切酶和连接酶,仅依赖噬菌体编码的重组酶蛋白,具有同源序列短(40~50 bp)[8],重组效率高[9-10],适用范围广和操作策略灵活的优势。
尽管研究人员很早就开始应用大肠杆菌重组工程系统进行基因操作,然而对于分枝杆菌;由于其同源重组率较低,且生长缓慢,使得针对它的同源重组操作较为困难。最早有人尝试直接利用大肠杆菌的重组工程系统进行结核分枝杆菌的基因置换,但由于分枝杆菌基因组的高 GC含量,导致大肠杆菌的这套系统不能正常行使功能。直到最近,Julia C van Kessel和 Graham F Hatfull在一种名为Che9c的分枝杆菌噬菌体中发现了类似RecE和Rec T蛋白功能的同源物——gp60和 gp61[7],它们的序列同源性接近30%,将这两个基因构建到载体pJV 53上,并导入结核分枝杆菌细胞内,研究基因置换效率,发现在这套系统的介导下,重组效率大大提高。
与以往使用的分枝杆菌重组质粒不同,利用pJV 53质粒编码的重组工程系统在进行基因打靶时,所需靶基因的同源片段长度在500 bp左右就可以达到很好的效果,本研究所使用的目的基因上下游同源片段长度为600 bp左右,即可将构建的敲入片段重组入耻垢分枝杆菌基因组中,远低于传统基因敲除所要求的超过1 000 bp的同源片段长度;且在操作中,这种方法无需使用任何特定的限制性酶切位点,即可在分枝杆菌体内完成重组过程,省去了体外酶切、连接等步骤,并能得到较高的重组效率。由此可见,这套分枝杆菌特有的重组工程系统具有传统方法无可比拟的优越性,为今后从事分枝杆菌的遗传操作带来了极大的便利。
[1]van Kessel J C,Hatfull G F.Recombineering in Mycobacterium tuberculosis[J].Nat Methods,2007,4(2):147-152.
[2]Rigaut G,Shevchenko A,Rutz B,et al.A generic protein purification method for protein comp lex characterization and proteomeexp lo ration[J].Nature biotechnology,1999,17(10):1030-1032.
[3]Puig O,Caspary F,Rigaut G,et al.The tandem affinity purification(TAP)method:a general procedure of protein complex purification[J].Methods,2001,24(3):218-229.
[4]Parish T,Stoker N G.Electroporation of mycobacteria[M].Methods in Molecular Biology:Mycobacteria Protocols,1998,101:129-144.
[5]Friedman D I,Court D L.Bacteriophage lambda:alive and well and still doing its thing[J].Current opinion in microbiology,2001,4(2):201-207.
[6]Poteete A R.What makes the bacteriophage lambda Red system useful for genetic engineering:molecular mechanism and biological function[J].FEMS microbiology letters,2001,201(1):9-14.
[7]Angrand P O,Daigle N,van der Hoeven F,et al.Simplified generation of targeting constructs using ET recombination[J].Nucleic acids research,1999,27(17):e16.
[8]Yu D,Ellis H M,Lee E C,et al.An efficient recombination system for chromosome engineering in Escherichia coli[J].Proceedings of the National Academy of Sciences of the United States of America,2000,97(11):5978-5983.
[9]Murphy KC,Campellone K G,Poteete A R.PCR-mediated gene replacement in Escherichia coli[J].Gene,2000,246(1-2):321-330.
[10]Court D L,Sawitzke J A,Thomason L C.Genetic engineering using homologous recombination[J].Annual review of genetics,2002,36:361-388.
Knocking TAP tag in the genome of Mycobacterium smegmatismc2155 by the recombineering system in Mycobacterium
(N ational Institute for Comm unicable D isease Control and Prevention,Chinese Center for Disease Control and Prevention/State Key Laboratory for Infectious D isease Prevention and Control,Beijing 102206,China)
ZHAO Li-li,XIA Qiang,ZHAO Xiu-qin,WAN Kang-lin
In order to knock the TAP tag in the genome of Mycobacterium smegm atis(M.smegm atis)mc2155 using the pJV 53 plasmid encoding recombineering system,pJV 53 plasmid was transformed into M.smegmatismc2155 to make the M.smegm atis mc2155 express recombinant proteins and p repare the electrocompetent cells.The tap gene was amp lified from pBS1479 plasmid and the hyg gene was amp lified from pSM T3 plasmid.The aasf(anti-anti-sigma factor)gene with AASF5 fragment(5’-noncoding region of aasf gene)and AASF3 fragment(3’-noncoding region of aasf gene)were amplified from the genome of M.smegm atismc2155.These four fragments were spliced together to form the knock-in fragment A 5THA 3 by overlap PCR.The constructed fragment was transformed into the electrocompetent cells to make it recombinant into the genome of M.smegmatismc2155.The recombinant effect was verified by PCR and DNA sequencing.The knock-in fragment of 3200bp was gotten by overlap PCR.It was confirmed by PCR and DNA sequencing that the A 5THA 3 fragment with TAP tag was knocked into the genome of M.smegm atismc2155 correctly.The TAP tag was successfully knocked into the genome of M.smegm atismc2155 and paved the way for further study of the functions of some target genes.
recombineering system;pJV 53 plasmid;tandem affinity purification(TAP)tag;knock-in fragment;overlap PCR;M ycobacterium smegmatis
R378.911
A
1002-2694(2011)06-0539-04
*国家“十一五”重大传染病防治科技重大专项“结核病传播模式研究”(2008ZX100/03-010-02)资助
万康林,Email:wankanglin@icdc.cn
1.中国疾病预防控制中心传染病预防控制所/传染病预防控制国家重点实验室,北京 102206;2.南华大学病原生物研究所,衡阳 421001
2011-01-26;
2011-03-03
