钠离子通道基因SCN1A突变及其相关癫综合征
2011-01-19许小菁张月华
许小菁 张月华

1 SCN1A基因与电压门控Na+通道
SCN1A基因定位于染色体2q24.3,长81 kb,有26个外显子,编码Na+通道α1亚单位。SCN1A基因属电压门控Na+通道编码基因家族中的一员,该家族还包括SCN2A、SCN3A、SCN7A和SCN9A基因[3]。
α1亚单位作为主体形成通道孔,由2 000个氨基酸组成,具有典型的4×6结构,由4个高度同源的结构域(D1~D4)组成,每个结构域含有6个跨膜区域(S1~S6,从左至右),N端和C端直接朝向细胞内部[4](图1)。
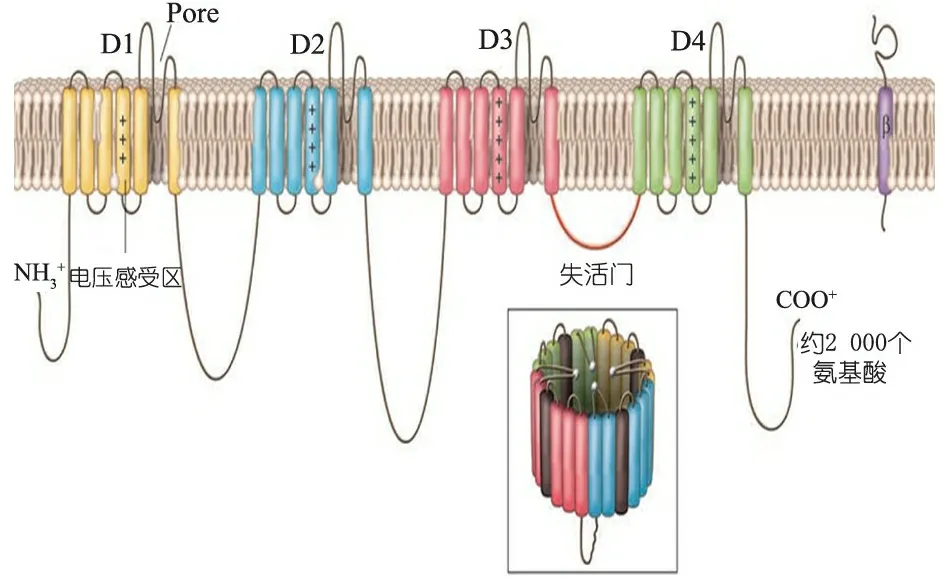
图1 Na+通道α1亚单位结构模式图[4]
S4区含5~8个带正电荷的氨基酸残基,称为“电压感受区”;S5和S6间的结构形成反向平行的β折叠衬于孔道内壁,称为“门孔区”,Na+经此区而由细胞膜外流入膜内;D3和D4间的细胞内连接环为“失活门”。
电压门控Na+通道存在于大多数可兴奋组织细胞膜上的跨膜大分子糖蛋白,是一种电压敏感性通道,主要具有触发动作电位作用。通道内部传感结构能够感知细胞膜上的微小电位变化,同时也能被S4跨膜区域上的正电荷残基感知,产生去极化,使S4区域的碱性残基朝向细胞外侧。这些变化反过来可引发“活化门”的构象重排,使离子孔传导时间缩短,仅为数毫秒,离子孔即关闭成为“失活门”。任何细微的电压变化对于门控通道的影响都是重大的,进而影响到细胞的兴奋性[2]。
2 SCN1A基因突变相关癫综合征
Escayg等[14]首次报道在GEFS+家系中发现SCN1A基因突变。5%~10%的GEFS+家系存在SCN1A基因突变[15,16]。迄今为止,在GEFS+家系中已发现42种SCN1A基因突变类型,且均为错义突变(http://www.molgen.ua.ac.be/SCN1AMutations/ Mutations/ Default.cf-m)。
GEFS+家系具有遗传异质性,虽然SCN1A基因是GEFS+最常见的致病基因;但SCN1B和氨基丁酸受体γ2亚单位基因(GABRG2)基因突变也与GEFS+家系发病有关[12,16,17];SCN2A和GABRD基因突变也分别见诸于一个GEFS+家系报道[18,19]。

表1 20个SCN1A 基因突变GEFS+家系表型分析[14~16,20~33]
近年的研究结果显示,约80%的Dravet 综合征患儿具有SCN1A基因突变,基因突变类型也达300余种,包括截断突变、错义、无义、碱基缺失及重复等,其中超过一半的患者因无义和框移突变导致蛋白质的截断[34]。这些突变广泛分布于整个亚单位蛋白的C端至N端,包括形成Na+通道孔的重要功能区(S5~6)[36]。SMEB和SMEI患儿的SCN1A基因突变率无明显区别,从分子遗传学的水平也证实两者无本质区别,故两者均称为Dravet综合征。Fujiwara等[37]在10例ICEGTC患儿中发现7例均为SCN1A基因错义突变,与SMEI、SMEB中SCN1A基因突变率接近。Harkin等[38]在2例ICEGTC患儿中发现了1例缺失突变(Q1914fsX1943)。
PCR-DNA测序未发现SCN1A基因突变的病例,采用多重连接依赖的探针扩增技术(MLPA) 已发现SCN1A基因的片段缺失或重复,从单一外显子至整个基因的缺失,在Dravet综合征中的阳性率为10%~15%[39~42]。采用MLPA的方法还可发现染色体的微缺失,再通过比较基因组杂交和荧光原位杂交的方法验证缺失位置和大小,并且能发现SCN1A邻近基因的缺失。
有研究[46]发现,8%的Dravet综合征病例有SCN9A基因突变, 9例SCN9A基因突变中有6例同时存在SCN1A基因突变,分析认为SCN9A可能是SCN1A的修饰调节基因。 Depienne等[47]发现SCN1A基因突变阴性的SEMI患儿中,15%的女性患儿有原钙黏蛋白基因PCDH19 突变, 该基因定位于染色体Xq22。提示PCDH19 基因与Dravet综合征女性患者致病高度相关,此外有1例男性SMEI患者为PCDH19 基因突变嵌合体。
SIMFE临床特点:SIMFE可被归入CFE。SIMFE临床表现为1岁以内早发的多变的部分性发作;EEG以大量多灶性放电为主,不伴全面性或双侧同步化放电;随病情发展出现不同程度的智力发育落后。已在5例患者中发现3种SCN1A基因突变(F575fsSX48、F1543S和F1543S)[38]。
CGE临床特点:可表现为多种发作形式,智力倒退,EEG有全导棘慢波。已发现6例CGE患儿SCN1A基因的错义突变(T226M、A395P、V422E、S626G、M973V和IVS15+1G→T)[38]。
3 SCN1A基因型与表型相关性分析
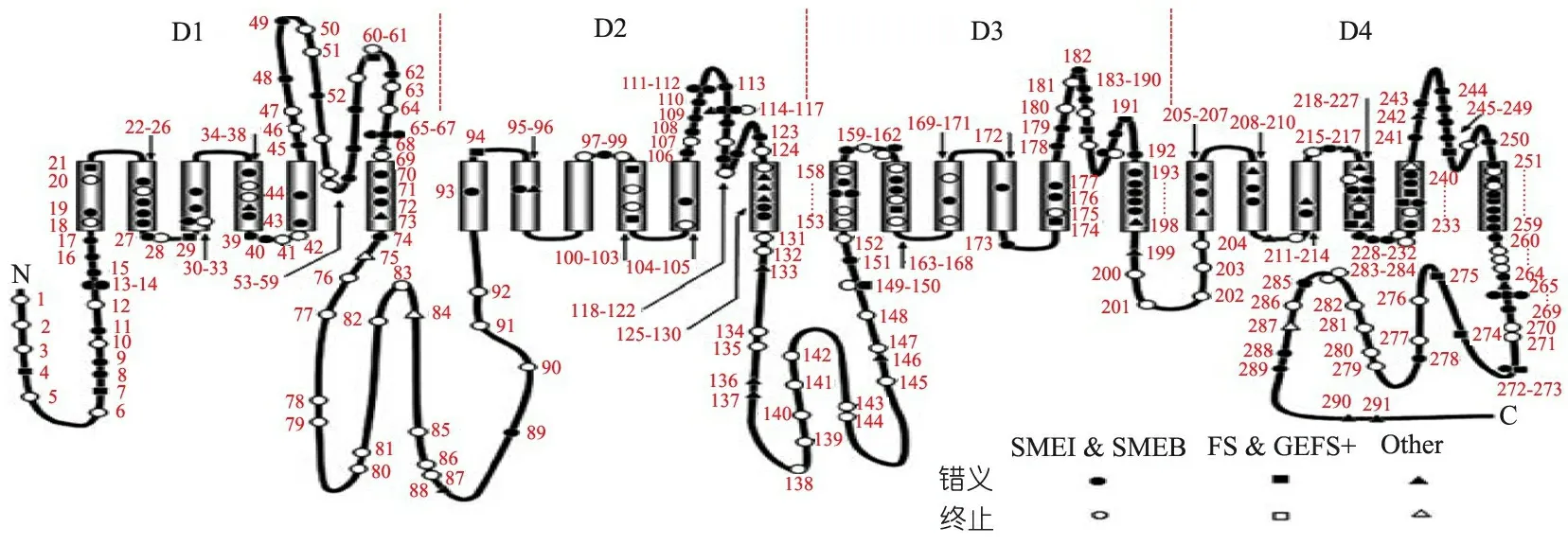
图2SCN1A基因突变位点分布(不包括大片段缺失和重复)[2]
4 SCN1A基因突变功能研究
电压门控Na+通道负责控制神经元和其他可兴奋细胞动作电位的上升支[4]。Na+通道α亚单位蛋白编码基因SCN1A的突变可导致Na+通道的功能获得,如持续的Na+内流,也可导致Na+通道的功能缺失,如Na+通道密度减少,激活与失活的电压依赖性改变[62]。
在SCN1A基因突变功能研究中,GEFS+中SCN1A基因突变常导致Na+通道失活[65]。Sugawara等[69]将Dravet综合征SCN1A基因的错义突变和无义突变(G979R、N985I、F1831S、R712X,R1407X、R1892X)转入HEK293细胞,通过膜片钳功能研究发现,细胞Na+电流明显减少,通道功能缺失;其中截断突变R712X、R1407X和R1892X可引起单倍体功能不足,通道表达量下降,导致通道活性的完全缺失。Dravet综合征相关的SCN1A基因突变R1648C与F1661S在体外突变模型中功能获得和缺失同时存在[68]。
5 SCN1A基因突变筛查的意义
[1]Mulley JC, Scheffer IE, Petrou S, et al. SCN1A mutations and epilepsy. Hum Mutat ,2005,25(6):535-542
[2]Lossin C.A catalog of SCN1A variants.Brain Dev,2009,31(2):114-130
[3]Malo MS, Blanchard BJ, Andresen JM ,et al. Localization of a putative human brain sodium channel gene (SCN1A) to chromosome band 2q24. Cytogenet Cell Genet, 1994,67(3):178-186
[4]Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest,2005, 115(8):2010-2017
[5]McEwen DP, Meadows LS, Chen C,et al. Sodium channel beta1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J Biol Chem,2004,279(16):16044-16049
[6]Isom LL. The role of sodium channels in cell adhesion. Front Biosci ,2002,7(1):12-23
[7]Patton DE, Isom LL, Catterall WA, et al. The adult rat brain beta 1 subunit modifies activation and inactivation gating of multiple sodium channel alpha subunits.J Biol Chem, 1994,269(26):17649-17655
[8]Fozzard HA, Hanck DA. Structure and function of voltage dependent sodium channels:comparison of brain II and cardiac isoforms.Physiol Rev ,1996,76(3):887-926
[9]Meadows LS,Malhotra J,Loukas A, et al. Functional and biochemical analysis of a sodium channel beta1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1.J Neurosci,2002,22(24):10699-10709
[10]Gorter JA,van Vliet EA,Lopes da Silva FH,et al. Sodium channel beta1-subunit expression is increased in reactive astrocytes in a rat model for mesial temporal lobe epilepsy. Eur J Neurosci ,2002,16(2):360-364
[11]Chen C,Bharucha V,Chen Y, et al. Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc Natl Acad Sci USA, 2002,99(24):17072-17077
[12]Wallace RH,Wang DW,Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet, 1998,19(4):366-370
[13]Scheffer I, Berkovic S. Generalized epilepsy with febrile seizures plus: a genetic disorder with heterogeneous clinical phenotypes. Brain, 1997,120(Pt3):479-490
[14]Escayg A, MacDonald BT,Meisler MH,et al. Mutations of SCN1A, encoding a neuronal sodium channel,in two families with GEFS+2. Nat Genet,2000,24(4):343-345
[15]Escayg A,Heils A,MacDonald BT,et al. A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus - and prevalence of variants in patients with epilepsy. Am J Hum Genet,2001, 68(4):866-873
[16]Wallace RH,Scheffer IE,Barnett S,et al. Neuronal sodium-channel a1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet,2001, 68(4):859-865
[17]Baulac S, Huberfeld G, Gourfinkel-An I, et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet, 2001,28(1):46-48
[18]Sugawara T, Tsurubuchi Y, Agarwala KL, et al. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci USA, 2001,98(11):6384-6389
[19]Dibbens LM, Feng HJ, Richard MC, et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet, 2004,13(13):1315-1319
[20]Baulac S, Gourfinkel-An I, Picard F, et al. A second locus for familial generalised epilepsy with febrile seizures plus maps to chromosome 2q21-q33. Am J Hum Genet, 1999,65(4):1078-1085
[21]Abou-Khalil B, Ge Q, Desai R, et al. Partial and generalised epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology, 2001,57(12):2265-2272
[22]Sugawara T, Mazaki-Miyazaki E, Ito M, et al. Na(v)1.1 mutations cause febrile seizures associated with afebrile partial seizures. Neurology, 2001,57(4):703-705
[23]Moulard B, Guipponi M, Chaigne D, et al.Identification of a new locus for generalized epilepsy with febrile seizures plus (GEFS+) on chromosome 2q24-q33. Am J Hum Genet, 1999,65(5):1396-1400
[24]Sun HH, Zhang YH, Liang JM, et al. SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalized epilepsy with febrile seizures plus. J Hum Genet, 2008,53(8):769-774
[25]Nagao Y, Mazaki-Miyazaki E, Okamura N, et al. A family of generalized epilepsy with febrile seizures plus type 2-a new missence mutation of SCN1A found in the pedigree of several patients with complex febrile seizures. Epilepsy Res, 2005,63(2-3):151-156
[26]Barela AJ, Waddy SP, Lickfett JG, et al. An epilepsy mutation in the sodium channel SCN1A that decreases channel excitability. J Neurosci, 2006,26(10):2714-2723
[27]Colosimo E, Gambardella A, Mantegazza M, et al. Electroclinical features of a family with simple febrile seizures and temporal lobe epilepsy associated with SCN1A loss-of-function mutation. Epilepsia ,2007,48(9):1691-1696
[28]Mahoney K, Moore SJ, Buckley D, et al. Variable neurological phenotype in a GEFS+ family with a novel mutation in SCN1A. Seizure,2009,18(7):492-497
[29]Dimova PS, Yordanova I, Bojinova V, et al. Generalized epilepsy with febrile seizures plus: Novel SCN1A mutation. Pediatr Neurol, 2010,42(2):137-140
[30]Fendri-Kriaa N, Kammoun F, Rebai A, et al. Genetic screening of two Tunisian families with generalized epilepsy with febrile seizures plus. Eur J Neurol,2009,16(6):697-704
[31]Herini ES, Gunadi, Harahap ISK, et al. Generalized epilepsy with febrile seizures plus (GEFS+) spectrum: Clinical manifestations and SCN1A mutations in Indonesian patients. Epilepsy Res, 2010,90(1-2):132-139
[32]Pineda-Frujillo N, Carrizosa J, Cornejo W, et al. A noval SCN1A mutation associated with severe GEFS+ in a large South American pedigree. Seizure, 2005,14(2):123-128
[33]Osaka H, Ogiwara I, Mazaki E, et al. Patients with a sodium channel alpha 1 gene mutation show wide phenotypic variation. Epilepsy Res, 2007,75(1):46-51
[34]Mullen SA, Scheffer IE. Translational Research in Epilepsy Genetics: sodium channels in man to interneuronopathy in mouse. Arch Neurol,2009,66(1):21-26
[35]Oguni H, Hayashi K, Awaya Y,et al. Severe myoclonic epilepsy in infants: a review based on the Tokyo Women's Medical University series of 84 cases. Brain Dev, 2001,23(7):736-748
[36]Catterall WA. From ionic currents to molecular mechanisms:the structure and function of voltage gated sodiumchannels. Neuron, 2000, 26(1):13-25
[37]Fujiwara T, Sugawara T, Mazaki-Miyazaki E,et al. Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain ,2003,126(Pt3):531-546
[38]Harkin LA, McMahon JM, Iona X,et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain, 2007,130(Pt3):843-852
[39]Marini C, Mei D, Temudo T,et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia,2007, 48(9):1678-1685
[40]Mulley JC, Nelson P, Guerrero S, et al. A new molecular mechanism for severe myoclonic epilepsy of infancy:exonic deletions in SCN1A. Neurology,2006, 67(6):1094-1095
[41]Wang JW, Kurahashi H, Ishii A, et al. Microchromosomal deletions involving SCN1A and adjacent genes in severe myoclonic epilepsy in infancy. Epilepsia, 2008,49(9):1528-1534
[42]Sun HH, Zhang YH, Liu XY, et al. Analysis of SCN1A mutation and parental origin in patients with Dravet syndrome. J Hum Genet, 2010,55(7):421-427
[43]Nabbout R, Gennaro E, Dalla Bernardina B, et al.Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy.Neurology,2003,60(12):1961-1967
[44]Depienne C, Arzimanoglou A, Trouillard O, et al.Parental mosaicism can cause recurrent transmission of SCN1A mutations associated with severe myoclonic epilepsy of infancy.Hum Mutat,2006,27(4):389
[45]Heron SE, Scheffer IE, Iona X, et al.De novo SCN1A mutations in Dravet syndrome and related epileptic encephal-opathies are largely of paternal origin.J Med Genet,2010,47(2):137-141
[46]Singh NA, Pappas C, Dahle EJ,et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome.PLoS Genet,2009,5(9):e1000649
[47]Depienne C, Bouteiller D, Keren B,et al.Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females.PLoS Genet,2009,5(2):e1000381
[48]Okumura A, Kurahashi H, Hirose S, et al. Focal epilepsy resulting from a de novo SCN1A mutation. Neuropediatrics, 2007,38(5): 253-256
[49]Doose H. Das akinetische petit mal. Arch Psychiatr Nervenkr, 1964,205:637-654
[50]Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia,1989,30(4):389-399
[51]Scheffer IE, Wallace R, Mulley JC, et al.Clinical and molecular genetics of myoclonic-astatic epilepsy and severe myoclonic epilepsy in infancy (Dravet syndrome).Brain Dev, 2001,23(7):732-735
[52]Wallace RH, Hodgson BL,Grinton BE,et al. Sodium channel alpha1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology,2003,61(6):765-769
[53]左启华,主编.小儿神经系统疾病.第2版.北京:人民卫生出版社,2002.296-298
[54]Livingston JH,Cross JH, Mclellan A, et al.A novel inherited mutation in the voltage sensor region of SCN1A is associated with Panayiotopoulos syndrome in siblings and generalized epilepsy with febrile seizures plus. J Child Neurol, 2009, 24(4): 503-508
[55]Mantegazza M, Gambardella A, Rusconi R, et al. Identification of an Nav1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc Natl Acad Sci USA,2005,102(50):18177-18182
[56]Colosimo E, Gambardella A, Mantegazza M,et al. Electroclinical features of a family with simple febrile seizures and temporal lobe epilepsy associated with SCN1A loss-of-function mutation. Epilepsia, 2007,48(9):1691-1696
[57]Weiss LA, Escayg A, Kearney JA, et al. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry, 2003,8(2):186-194
[58]Gargus JJ, Tournay A. Novel mutation confirms seizure locus SCN1A is also familial hemiplegic migraine locus FHM3. Pediatr Neurol ,2007,37(6):407-410
[59]Dichgans M, Freilinger T, Eckstein G,et al. Mutation in the neuronal voltagegated sodium channel SCN1A in familial hemiplegic migraine.Lancet, 2005,366(9483):371-377
[60]Vanmolkot KR, Babini E, de Vries B,et al. The p.L1649Q mutation in the SCN1A epilepsy gene is associated with familial hemiplegic migraine:genetic and functional studies. Hum Mutat,2007, 28(5):522
[61]Ceulemans BP, Claes LR, Lagae LG. Clinical correlations of mutations in the SCN1A gene: from febrile seizures to severe myoclonic epilepsy in infancy. Pediatr Neurol,2004, 30(4):236-243
[62]Sun HH(孙慧慧),Zhang YH.Progress in molecular genetics in severe myoclonic epilepsy of infancy.Chin J Pediatr(中华儿科杂志),2008,46(2):157-160
[63]Wang W, Takashima S, Segawa Y, et al.The developmental changes of Na(v)1.1 and Na(v)1.2 expression in the human hippocampus and temporal lobe.Brain Res,2011,1389:61-70
[64]Alekov AK, Rahman MM, Mitrovic N, et al. Enhanced inactivation and acceleration of activation of the sodium channel associated with epilepsy in man. Eur J Neurosci, 2001,13(11):2171-2176
[65]Lossin C, Wang DW, Rhodes TH,et al. Molecular basis of an inherited epilepsy. Neuron, 2002,34(6):877-884
[66]Spampanato J, Escayg A, Meisler AH, et al.Generalized epilepsy with febrile seizures plus type 2 mutation W1204R alters voltage-dependent gating of Na(v)1.1 sodium channels. Neuroscience,2003,116(1):37-48
[67]Vanoye CG, Lossin C, Rhodes CH, et al Single-channel properties of human Nav1.1 andmechanism of channel dysfunction in SCN1A associated epilepsy. J Gen Physiol, 2006,127(1):1-14
[68]Rhodes TH, Lossin C, Vanoye CG, et al. Noninactivating voltagegated sodium channels in severe myoclonic epilepsy of infancy. Proc Natl Acad Sci USA, 2004,101(30):11147-11152
[69]Sugawara T,Tsumbuchi Y,FujiwaraT,et al. Navl.1channels with mutations of severe myoclonic epilepsy in infancy display attenuated currennts.Epilepsy Res,2003,54(2-3):201-207
[70]Gambardella A, Marini C.Clinical spectrum of SCN1A mutations.Epilepsia,2009,50(S5):20-23
[71]Ottman R, Hirose S, Jain S, et al. Genetic testing in the epilepsies-report of the ILAE Genetics Commission. Epilepsia, 2010,51(4):655-670
