作用于不同活性位点的单胺氧化酶抑制剂
2011-01-14郭跃平郑瑞华梁世君胡基埂肖延铭
郭跃平 郑瑞华 梁世君 胡基埂 肖延铭
(1.桐庐县质量计量监测中心,浙江 杭州 311500;2.浙江工业大学,浙江 杭州 310014)
0 引言
单胺氧化酶(monoamine oxidase,MAO,EC1.4.3.4)全名为单胺-氧氧化还原酶,是一种催化氧化一些生物体产生的胺包括神经传递素多巴胺、去肾上腺素(NE)、血清素(5-HT)、酪胺、苯乙胺(PEA)以及神经毒素1-甲基-4-苯基-1,2,3,6-四氢嘧啶(MPTP)等的酶(图1)。单胺氧化酶是一种位于线粒体膜外的整合蛋白,根据底物选择性和对抑制剂的灵敏度,可以分为两种亚型:MAO-A和MAO-B。其中单胺氧化酶A对底物去肾上腺素(NE)、血清素(5-HT)、多巴胺(DA)和抑制剂clorgyline(图1)具有高亲和性;而单胺氧化酶B则对苯乙胺(PEA)、苯甲胺和抑制剂(R)-deprenyl(图1)具有高亲和性[1]。研究表明MAO-A和MAO-B是两种截然不同的蛋白质,cDNA克隆试验显示MAO-A由526个氨基酸组成,分子量为59700,而MAO-B由520个氨基酸组成,分子量为58800[2]。在人体中除了血小板外大多数组织中单胺氧化酶A和B都存在,其中肝脏和胎盘中含量较高,脾中含量最低。MAO-A在胎盘、肺和小肠中含量占优势而MAO-B则在心肌中占优势[3-4]。人大脑中也普遍存在MAO,其中MAO-A多位于多巴胺能的神经元细胞,而MAO-B则多位于含血清素的神经元细胞中[5-7]。
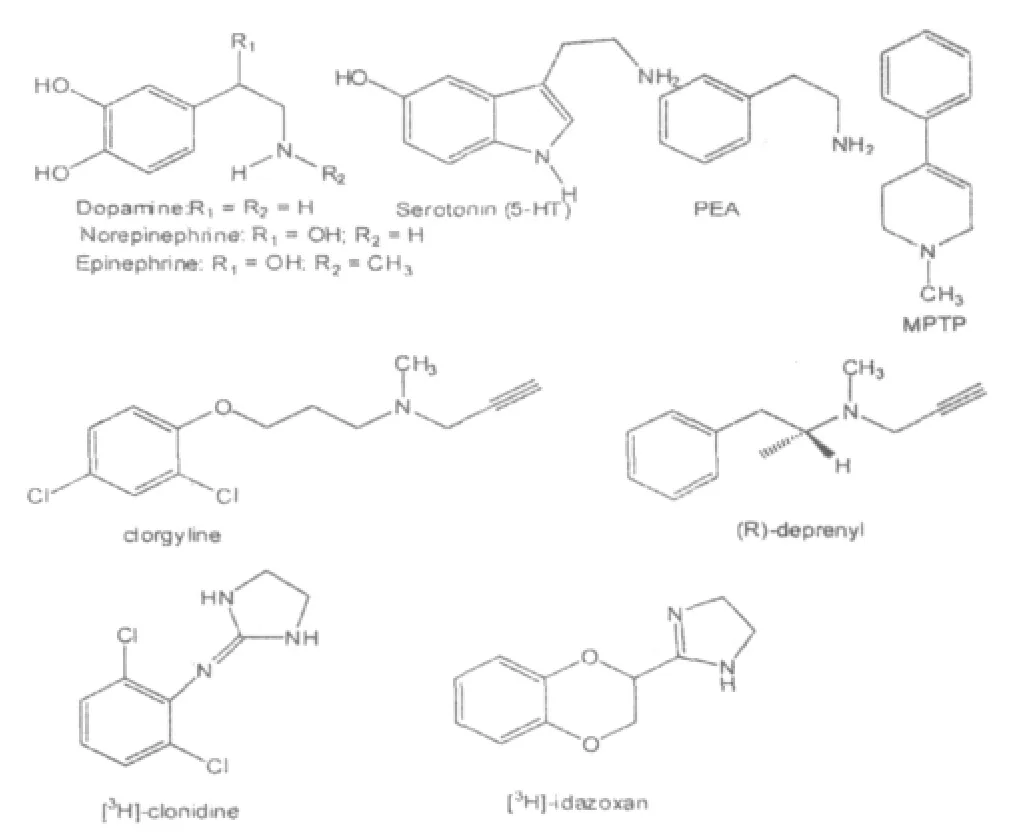
图1 单胺氧化酶抑制剂
1 单胺氧化酶结构与功能
光谱学试验表明单胺氧化酶的二级结构可分为四个区域:
(1)ADP键结合区域(residues 6~43);
(2)假设的底物结合区域(residues 178~221);
(3)FAD共价键辅酶所在区域(residues 350~458);
(4)C端(residues 491~511)和线粒体膜结合的α-螺旋结构区域[8-9](图2)。

图2
图2左侧MAO-B的结构,包括三个功能区域:红色为底物作用区域其中青色为两个空腔,外侧的是入口空腔,内侧为底物结合的空腔;蓝色显示的是核黄素结合的区域,其中黄色表示FAD分子;绿色为C-端螺旋状区域,它使酶结合到线粒体膜上。右侧为MAO-A的结构,区域颜色与左侧一致[10]。
用拉扎贝胺(N-[2-aminoethyl]-5-chloro-2-pyridinecarboxamide)作为假设的抑制剂做定向部位突变试验显示MAO-B中氨基酸残基组氨酸382(His382)和苏氨酸158(Thr158)是酶起催化作用的必要部分,而MAO-A中的苯丙氨酸208(Phe208)和MAO-B中的异亮氨酸199(Ile199)则是决定底物特异性的关键部分[11-13]。Shih等人[14]发现在酶的活性位点存在两个半胱氨酸的残基与催化作用有密切的关系。Ramsay等人[15]发现MAO-A和MAO-B的活性位点中都含有一个氧化还原二硫化物的活性位点,因此单胺氧化酶可能被描述为二硫化物氧化还原酶。通过转移和替换酶中的一些区域的试验是我们越来越多地了解酶的结构及其作用,许多已得到大家的共识。
虽然单胺氧化酶的两种同工酶对底物和抑制剂的选择性不同,但它们都能催化氧化一级、二级、三级胺。Ramsay等人[16]研究提出了单胺氧化酶催化作用的可能机制,MAO中的FAD氧化一级,二级,三级胺脱氨基生成相应的醛和自由胺,同时生成过氧化氢(图3)。电子从氮到氧的转移形式目前还没有达成共识,主要有三种观点:⑴单电子转移(SET);⑵氢原子转移(HAT);⑶亲核或极性机制。

图3
图3是MAO氧化脱氨基的反应途径,底物在MAO的作用下生成相应的醛,通常情况迅速被脱醛酶(ADH)氧化生成羧酸,排出体外。同时FADFADH2循环产生的过氧化氢在过氧化氢酶的作用下失活。
2 MAO抑制剂
自从偶然发现抗结核药异烟肼是一种潜在的MAO抑制剂以来,对MAO抑制剂的研究便成为了热点。异烟酰异丙肼成为第一个MAO抑制剂,因发现这类含有肼结构的MAO抑制剂同时能使P450s失活而导致肝脏毒性;又研究了以苯环丙胺为代表的非肼类的MAO抑制剂,这类可称为第二代,但因为其能引发高血压,便限制了它的应用。近年来可逆的选择性的MAO抑制剂成为了研究方向。
2.1 作用于"Aromatic Cage"的单胺氧化酶抑制剂
单胺氧化酶底物结合位点由正面的辅酶FAD的异咯嗪环和酶的两个近似垂直氨基酸残基酪氨酰构成(图4)[17],形象的称为"Aromatic Cage"。

图4
图4是MAO-B中的"Aromatic Cage",A是"Aromatic Cage"的俯视图,包括核黄素的环和Y398和Y435,核黄素环倾斜于平面大约30°虚线表示" aromatic cage"的中心面;B是两个酪氨酰残基的侧面图,碳原子用绿色表示,氮原子用蓝色表示,氧原子用红色表示[17]。
研究显示用苯丙氨酸替换人体MAO-A中的"Aromatic Cage"的两个酪酰胺残基(Y407和Y444)会明显影响酶的催化活性和对底物的特异性。通过结构数据的分析可以得出"Aromatic Cage"的作用是确保底物中胺的部分可以靠近FAD的活性部位,同时影响电子的转移,提高催化活性。因此,如果一些芳环类似物占据酶的"Aromatic Cage"部分,阻止底物接近FAD的活性部位就会达到抑制酶活性的目的,根据这一机制人们设计合成了许多的单胺氧化酶抑制剂。化合物3进入MAO-A的活性位点,苯环被置入"Aromatic Cage",通过和酪氨酸(Tyr407和Tyr444)π-π键叠加以及和FAD的芳环的T型π-π键重叠形成静电作用,是MAO-A的选择性抑制剂,而在MAO-B中因构型的影响使苯环不能和"Aromatic Cage"形成有效的重叠而抑制活性很低。化合物2在MAO-B中能形成有效的π-π键重叠,因而是MAO-B的选择性抑制剂。化合物4~10(图5)用炔基和环己基替换苯基使得选择性降低[18]。化合物12的吲哚环进入MAO-A中的"Aromatic Cage"表现出对MAO-A强选择性和抑制活性,化合物13因N上的甲基不能和FAD形成氢键,活性降低[19]。化合物11、14和15有很强的抑制活性但选择性弱。

图5 作用于"Aromatic Cage"的单胺氧化酶抑制剂
2.2 作用于I2-IBS活性位点的单胺氧化酶抑制剂
1984年Bousquet等人[21]发现抗高血压药物"可乐定"([2-(2,6-二氯苯基)亚氨基]咪唑烷)为咪唑活性位点(IBS)的底物[20],后来发现根据对不同配位基相互作用的不同咪唑活性位点可以分为三类:⑴I1型咪唑活性位点(I1BS)能与[3H]-可乐定和[3H]-咪唑克生(图1)紧密结合,分布在突触质膜,被认为是抗高血压药物的作用靶点;⑵I2型咪唑活性位点(I2BS)只能与[3H]-咪唑克生紧密结合外围和中心组织的线粒体膜外,后来研究发现I2BS的底物也是单胺氧化酶抑制剂;⑶I3型咪唑活性位点(I3BS),药理学家在胰脏β细胞和胰岛分泌物中发现一种非典型受体被推断为I3型咪唑活性位点(I3BS)。
I2BS与单胺氧化酶的关系引起了科学家的兴趣,许多咪唑活性位点(IBS)的底物也会与单胺氧化酶发生作用,蛋白质水平的研究发现在单胺氧化酶中含有缩氨酸,而缩氨酸的结构常出现在咪唑活性位点(IBS)的底物中。实验发现I2底物2-(2-benzofuranyl)-2-imidazoline(2-BFI)是单胺氧化酶A的抑制剂。同时"molecule dock"实验发现,2-BFI和[3H]-咪唑克生在单胺氧化酶中处于同一区域,基本由Tyr69,Tyr197,Phe208,Tyr407,Phe352,Tyr444组成。
随着研究的深入,许多含有咪唑结构的单胺氧化酶抑制剂被合成出来,如化合物22和23(图6)。其中许多将为我们进一步研究咪唑类单胺氧化酶抑制剂提供思路,化合物21在呋喃环的一侧为芳环,一侧为咪唑环[22],观察许多的单胺氧化酶抑制剂,我们会发现其实单胺氧化酶抑制剂可以分为三个部分:两侧为活性部分,中间为连接部分。这些结构的认识会为对我们研究单胺氧化酶抑制剂具有非常重要的作用。

图6 作用于I2-IBS活性位点的单胺氧化酶抑制剂
2.3 作用于单胺氧化酶通道的抑制剂
近年来单胺氧化酶的晶体结构已经阐明,发现从表面到酶的活性中心存在一条重要通道,这条通道有两个空腔组成,分别为入口空腔("entrance cavity")和底物空腔("substrate cavity")。这两个空腔既是彼此分离的又是密切连接的,在MAO-B中的由Ile-199控制,我们形象的称为"gate",而在MAO-A中是Phe208[23]。试验表明"gate"的结构关系到抑制剂的选择性。化合物24占据底物空腔氨基茚满环使得Ile-199处于开放状态,相对于MAO-A,MAO-B底物空腔能容纳体积更大的基团[24]。化合物25和26能横跨入口空腔和底物空腔,其中化合物23的羟基结合到FAD上[25]。所以作用于MAO通道的这一类抑制剂一般包括三个部分:⑴亲脂性基团使抑制剂能结合到入口空腔;⑵中间连接部分,关系到抑制剂的选择性;⑶富电子基团,因为线粒体膜带阴离子方便胺定向进入酶,所以含有富电子基团也有利于抑制剂进入酶[26]。
化合物28和29的苯环作用于"entrance cavity",而香豆素环作用于"substrate cavity"。化合物30使28和29的衍生物活性增强[27]。化合物31是MAOB的选择性抑制剂,其中的杂环决定其选择性[28]。化合物34的左边的带甲基的环与"substrate cavity"中的Phe177,Leu176,Phe173,和Ile325有强的作用,右边两个环占据"entrance cavity",中间的杂原子会和MAO-B中的氨基酸残基形成氢键(Cys172),而在MAO-A中没有形成氢键,所以对MAO-B有较好选择性[29]。化合物35的氟代苯环占据"entrance cavity",左侧部分作用于"substrate cavity",酰胺部分和FAD附近的结构性水分子形成氢键。(R)-36对MAO-B选择性增加[30]。化合物37和38的苯环插入Tyr398和Tyr435之间,腈基延伸到"entrance cavity",和Cys172,Gln206,Ph168以及Ile199等氨基酸残基的静电作用增加了其在"entrance cavity"的稳定性[31]。化合物41是MAO-A的选择性抑制剂,其选择性是化合物40的100多倍,说明链的长度对活性有影响,化合物39是40的衍生物[32]。

图7 作用于单胺氧化酶通道的抑制剂
2.4 其他类
2.4.1 炔类
化合物51的炔胺结构和FAD的N5作用形成稳定结构(图8),从而实现抑制作用。

图8 抑制剂与单胺氧化酶的作用机制
2.4.2 活性基团组合形成新的抑制剂
化合物52由具有活性的香豆素类抑制剂的衍生物和化合物49的类似物组成,形成一类新的MAO的抑制剂[33]。这类以已知具有抑制活性的结构组合形成的MAO抑制剂的方法,成为一种设计新抑制剂的重要方法,目前报道了许多这类抑制剂。化合物49和51的活性基团组合形成了化合物53[34],是MAO-A的高选择性抑制剂(图9)。

图9 设计性抑制剂的模型
3 结束语
在过去30多年的研究过程当中,大量的MAO抑制剂被合成出来,种类繁多,过去大多以化学结构分类,本文尝试以作用于酶的活性位点的不同进行分类。因数量众多,本文涉及到的抑制剂为具有代表性的抑制剂。对这些抑制剂的总结,不难发现具有选择性的可逆的MAO抑制剂将成为其发展的方向,因此对作用于不同活性位点的抑制剂的总结分析有助于我们认识和研究酶的结构和功能,同时也为我们抑制剂的研究提供思路,从而找到活性和选择性更高的抑制剂。
[1]Kalgutkar A S,Dalvie D K,Jr N C,et al.Interactions of nitrogen-containing xenobiotics with monoamine oxidase(MAO)isozymes A and B:SAR Studies on MAO substrates and inhibitors[J].Chemical Research in Toxicology,2001,9(14):1139-1162.
[2]Kuwahara T,Takamoto S,Ito A.Primary structure of rat monoamine-oxidase a deduced from cada and its expression in rat-tissues[J].Agric.Biol.Chem.,1990,54(1):253-257.
[3]O'Carroll A M,Anderson M C,Tobbia I,et al.Determination of the absolute concentrations of monoamine oxidase A and B in human tissues[J].Biochem.Pharmacol.,1989,38(6):901-905.
[4]Saura J,Nadal E,Van den Berg B,et al.Localization of monoamine oxidase in human peripheral[J].Life Sci.,1996,59(16):1341-1349.
[5]Thorpe L W,Westlund,K N,Kochersperger L M,et al.Immunocytochemical localization of monoamine oxidases A and B in human peripheral tissues and brain[J].J.Histochem.Cytochem.,1987,35(1):23-32.
[6]Bond P A,Cundall R L.Properties of monoamine oxidase(MAO)in human blood platelets,plasma,lymphocytes and granulocytes[J].Clin.Chim.Acta.,1977,80(2):317-326.
[7]Westlund,Karin N.The distribution of monoamine oxidases A and B in normal human brain[J].Neurol.Dis.Ther.,1994,21:1-19.
[8]Kim H,Sablin S O,Ramsay R R.Inhibition of monoamine oxidase A by b-carboline derivatives[J].Arch.Biochem.Biophys.,1997,37(1):137-142.
[9]Edmondson D E,Mattevi A,Binda C,et al.Structure and mechanism of monoamine oxidase[J].Curr.Med.Chem.,2004,11:1983-1993.
[10]Edmondson D E,Binda C,Mattevi A.Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B[J].Arch.Biochem.Biophys.,2007,464(2):269-276.
[11]Tsugeno Y,Ito A.A key amino acid responsible for substrate selectivity of monoamine oxidase A and B[J].J.Biol.Chem.,1997,272:14033-14036.
[12]Cesura A M,Gottowik J,Lang G,et al.Structurefunction relationships of mitochondrial monoamine oxidase A and B:Chimaeric enzymes and site-directed mutagenesis studies[J].J.Neural.Transm-Supp.,1998,52:189-200.
[13]Veselovsky,AV;Ivanov,AS;Medvedev,AE.Is one amino acid responsible for substrate specificity of monoamine oxidase A and B?[J].Biochemsitry-Moscow,1998,63(12):1441-1446.
[14]Wu H F,Chen K,Shin JC.Site-directed mutagenesis of monoamine oxidase-A and oxidase-B-role of cysteines[J].Mol.Pharmacol.,1993,43(6):888-893.
[15]Sablin S O,Ramsay R R.Monoamine oxidase contains a redox-active disulfide[J].J.Biol.Chem.,1998,273(23):14074-14076.
[16]Edmondson D E,Mattevi A,Binda C,et al.Structure and mechanism of monoamine oxidase[J].Curr.Med.Chem.,2004,11(15):1983-1993.
[17]Li M,Binda C,Mattevi A,et al.Functional role of the"aromatic cage"in human monoamine oxidase B:Structures and catalytic properties of Tyr435 mutant proteins[J].Current Medicinai Chemistry,2006,45(15):4775-4784.
[18]Regina G L,Silvestri R,Turini P,et al.New Pyrrole inhibitors of monoamine oxidase:synthesis,biological evaluation,and structural determinants of MAO-A and MAO-B selectivity[J].J.Med.Chem.,2007,50,922-931.
[19]La Regina G,Silvestri R,Gatti V,et al.Synthesis,structure-activity relationships andmolecular modeling studies of new indole inhibitors of monoamine oxidases A and B[J].Bioorg.Med.Chem.,2008,16(22):9729-9740.
[20]Jones T Z E,GiuratoL,GuccioneS,etal.Interactions of imidazoline ligands with the active site of purified monoamine oxidase A[J].Febs Journal,2007,274(6):567-1575.
[21]Dardonville C,Rozas I.Imidazoline binding sites and their ligands:An overview of the different chemical structures[J].Med.Res.Rev.,2004,24(5),639-661.
[22]Gentili F,Bousquet P,Brasili L,et al.Imidazoline binding sites(IBS)profile modulation:Key role of the bridge in determining I-1-IBS or I-2-IBS selectivity within a series of 2-phenoxymethylimidazoline analogues[J].J.Med.Chem.,2003,46(11):2169-2176.
[23]Hubalek F,Binda C,Khalil A,et al.Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors[J].J.Biol.Chem.,2005,280(16):15761-15766.
[24]Binda C,Hubalek F,Li M,et al.Binding of rasagiline-related inhibitors to human monoamine oxidases:a kinetic and crystallographic analysis[J].J.Med.Chem.,2005,48:8148-8154.
[25]Berg D,Zoellner K R,Ogunrombi M O,et al.Inhibition of monoamine oxidase B by selected benzimidazole and caffeine analogues[J].Bioorg.Med.Chem.,2007,15:3692-3702.
[26]Chimenti F,Bolascoa A,Mannaa F,et al.Synthesis,biological evaluation and 3D-QSAR of 1,3,5-trisubstituted-4,5-dihydro-(1H)-pyrazole derivatives as potent and highly selective monoamine oxidase a inhibitors[J].Curr.Med.Chem.,2006,13:1411-1428.
[27]Gnerre C,Catto M,Leonetti F,et al.Inhibition of monoamine oxidases by functionalized coumarin derivatives:biological activities,QSARs,and 3D-QSARs[J].J.Med.Chem.,2000,43:4747-4758.
[28]ChimentiF,FioravantiR,Bolasco A,etal.Monoamine oxidase isoform-dependent tautomeric influence in the recognition of 3,5-diaryl pyrazole inhibitors[J].J.Med.Chem.,2007,50:425-428.
[29]Chimenti F,Maccioni E,Secci D,et al.Selective inhibitory activity against MAO andmolecular modeling studies of 2-thiazolylhydrazone derivatives[J].J.Med.Chem.,2007,50:707-712.
[30]Leonetti F,Capaldi C,Pisani L,et al.Solid-phase synthesis and insights into structure-activity relationships of safinamide analogues as potent and selective inhibitors of type B monoamine oxidase[J].J.Med.Chem.,2007,50:4909-4916.
[31]Yelekci K,Karahan O,Toprakc M.Docking of novel reversible monoamine oxidase-B inhibitors:efficient prediction of ligand binding sites and estimation of inhibitors thermodynamic properties[J].J.Neural.Transm.,2007,114:725-732.
[32]Mai A,Artico M,Esposito M,et al.3-(1H-Pyrrol-1-yl)-2-oxazolidinones as reversible,highly potent,and selective inhibitors of monoamine oxidase type A[J].J.Med.Chem.,2002,45:1180-1183.
[33]Sunal1 S G,Yabanoglu S,Yesilada1 A,et al.Monoamine oxidase inhibitory activities ofnovel3,4-dihydroquinolin-(1H)-2-one derivatives[J].J.Neural.Transm.,2007,114:717-719.
[34]Hassan S Y,Khattab S N,Bekhitb A,et al.Synthesis of 3-benzyl-2-substituted quinoxalines as novel monoamine oxidase A inhibitors[J].Bioorg.Med.Chem.Lett.,2006,16:1753-1756.
