基于16S rRNA和recA香鱼鳗利斯顿氏菌的分离鉴定
2010-12-28王成义闫茂仓陈少波管敏鑫单乐州艾为明谢起浪蔡延马奔
王成义,闫茂仓,陈少波,管敏鑫,单乐州,艾为明,谢起浪,蔡延马奔
(1.温州医学院生命科学学院, 浙江 温州 325005;2.浙江省海洋水产养殖研究所,浙江 温州 325005;
3.浙江省近岸水域生物资源开发与保护重点实验室,浙江 温州 325005;4.Division of Human Genetics and Center for Hearing and Deafness Research, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio 45229, USA; 5.温州市工业科学研究院,浙江 温州 325028)
基于16S rRNA和recA香鱼鳗利斯顿氏菌的分离鉴定
王成义1,2,3,闫茂仓2,3,陈少波2,3,管敏鑫1,4,单乐州2,3,艾为明1,谢起浪2,3,蔡延马奔5
(1.温州医学院生命科学学院, 浙江 温州 325005;2.浙江省海洋水产养殖研究所,浙江 温州 325005;
3.浙江省近岸水域生物资源开发与保护重点实验室,浙江 温州 325005;4.Division of Human Genetics and Center for Hearing and Deafness Research, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio 45229, USA; 5.温州市工业科学研究院,浙江 温州 325028)
为研究引起香鱼(Plecoglossus altivelis)出血溃烂症病因及致病菌系统发育地位,本研究从患病香鱼的肝脏、肾脏及体表分离到11株病原菌(编号:X0901-X0911),运用常规细菌生理生化方法鉴定致病菌所属种类;运用16S rRNA基因、recA基因序列分析方法研究致病菌的系统发育地位。细菌生理生化鉴定结果表明:致病菌为鳗利斯顿氏菌,11株细菌生理生化结果相同,均为革兰氏阴性杆菌、氧化酶阳性、接触酶阳性、吲哚阳性、精氨酸脱羧酶阳性、精氨酸双水解酶阳性、硝酸盐还原阳性、甘露醇阳性、MR测定阳性;H2S阴性、V-P测定阴性等。根据16S rRNA基因、recA基因序列分别构建弧菌科常见细菌系统进化树,结果表明:致病菌与鳗利斯顿氏菌(Listonella anguillarum)均聚为一枝,聚类结果与细菌生理生化鉴定结果相符。致病菌与鳗利斯顿氏菌16S rRNA基因、recA基因的同源性分别为99.9%、99.8%。以recA基因构建的系统进化树的拓扑学结构与16S rRNA基因建树结果相类似。此外,与16S rRNA基因相比,recA基因在不同物种之间具有更高的鉴别能力,本研究表明recA基因适合作为弧菌科常见细菌物种间进化关系研究的标记。
香鱼; 鳗利斯顿氏菌; 分离; 鉴定
引 言
香鱼 (Plecoglossus altivelis) 隶属于鲑形目,香鱼科,香鱼属。是一种小型名贵经济鱼类,在当今国际市场上享有“淡水鱼之王”的美称,由于工业的发展及人为的滥捕,造成天然香鱼种群日益减少,目前已处于濒危的状态,被国家列为二级保护动物。1998年发布的《中国濒危动物红皮书》把香鱼列入易危动物[1]。在香鱼高密度人工养殖过程中极易出现病害,特别是在香鱼幼苗阶段易出现由鳗利斯顿氏菌(原:鳗弧菌vibrio anguillarum)、假单胞菌 (Pseudomonassp.)、链球菌 (Streptococcussp.)、柱状屈挠杆菌 (Flexibacter columnaris)、杀鱼巴斯德菌 (Past.Piscicida) 等引起的细菌性疾病,其中尤其以鳗利斯顿氏菌引发的出血溃烂病的影响最为严重[2]。
鳗利斯顿氏菌是一类革兰氏阴性菌,最早于1761年发现于欧洲,Bonaveri[3]将该病描述为红疫病,曾在18-19世纪意大利的海水养殖鳗鲡中流行,造成严重死亡。1909年,Bergman[4]从瑞典沿海养殖的患红疫病鳗鲡中分离到病原菌,并将其命名为鳗弧菌。MacDonnell和Colwell[5]在1985年将归属到利斯顿菌属(Listonella)。鳗利斯顿氏菌是一种条件致病菌,能够引起多种水产动物疾病,如:鲑鱼 (Stenodus leucichthys)、虹鳟 (Salmo grairdneri)、鳗鲡、香鱼、鲈鱼 (Lateolabrax japonicus)、鳕鱼 (Gadus callarisa)、大菱鲆 (Scophthatmus maximus)、牙鲆 (Paralichthys olivaceus)、大黄鱼 (Pseudosciaena crocea) 等[4,6-11]。
迄今为止,在细菌鉴定及系统发育地位研究方面,16S rRNA依然是最常用的分子工具。细菌分类学家普遍认为16S rRNA的同源性大于97.5%的细菌可视为同种[12],然而弧菌科许多不同种细菌的同源性大于97.5%[13],已经不能准确的确定待测菌株的系统分类学地位。在这种前提下,许多学者将视线转移到其他的持家基因作为系统分类学研究工具[14,15]。Zeigler[16]研究指出单个基因也可以很好的预测整个基因组的系统发育关系。recA基因是广泛存在于细菌中重要的持家基因,编码一种多功能蛋白质,其功能主要涉及:同源重组、DNA修复、SOS反应、单链DNA结合、DNA解螺旋、同源重组中寻找同源位点[17,18]。recA基因被用于研究Burkholderia属所有细菌的分类,证明是有效的鉴定工具[19]。Thompson等[14]利用recA做标记研究弧菌科细菌的系统发育关系,表明recA基因比16S rRNA具有更好的辨别能力,适合做为弧菌科细菌鉴定的工具。
本研究运用传统的细菌鉴定方法和16S rRNA基因和recA基因序列分析相结合的方法研究了引起香鱼出血溃烂病致病菌的系统分类学地位。目的在于研究香鱼的出血溃烂症的病因,为香鱼健康养殖提供理论基础。
1 材料与方法
1.1 材料
1.1.1 菌株、香鱼 菌株分离自典型的出血溃烂症状的香鱼;患病香鱼采自乐清市某香鱼育苗场,健康香鱼来自浙江省海洋水产养殖研究所清江试验场。
1.1.2 试剂 PCR扩增试剂盒、DL2000分子量标准、UNIQ-10柱式细菌基因组DNA抽提试剂盒(SK1202)均购自上海生工生物工程技术服务有限公司,细菌鉴定管购自杭州天和微生物有限公司。
1.2 方法
1.2.1 细菌的分离鉴定 取患出血病濒临死亡的香鱼,用70%酒精消毒体表,用无菌的剪刀剪破病鱼的体腔壁,在无菌条件下挑取肝胰脏、肾脏接种于ZOBELL 2216E和TCBS培养基,将平板置于30℃条件下培养24 h挑取形态一致的优势菌落,进一步划线纯化,经纯化培养后转接到ZOBELL 2216E海水斜面
培养基上,于4℃保存、备用。
1.2.2 人工感染试验 选取纯化后的4株代表细菌用于人工感染试验,方法是将供试菌株分别接种于ZOBELL 2216E海水培养基试管斜面,于30℃培养18 h,加入0.65%无菌的生理盐水冲洗菌落,配成菌悬液调整细菌浓度约为5.2×108个/mL,每尾香鱼胸腔注射0.2 mL菌液;另设对照组,每尾鱼注射相同剂量0.65%的无菌生理盐水,每组香鱼的数量为10尾。注射后观察香鱼的发病死亡情况,并对死亡香鱼及时剖检和致病菌的再次分离。
1.2.3 形态观察及生理生化鉴定 观察致病菌ZOBELL 2216E和TCBS培养基平板的菌落的形态特征,分别取11株致病菌的ZOBELL 2216E海水培养基试管斜面30℃的18 h培养物,制备相应的涂片,革兰氏染色,显微镜下观察个体形态、大小。按传统的细菌鉴定方法较系统地测定致病菌相关的理化特征指标。1.2.4 致病菌基因组DNA提取 将致病菌株分别接种于ZOBELL 2216E海水培养基试管斜面,于30℃培养18 h,加入0.65%无菌的生理盐水冲洗菌落,配置成为菌悬液。后依据UNIQ-10柱式细菌基因组DNA抽提试剂盒说明书所示方法提取致病菌的总DNA。基因组DNA于1%琼脂糖凝胶电泳检测模板的纯度和完整性。
1.2.5 16S rRNA基因和recA基因的PCR扩增、测序 使用primer premier5.0软件设计鳗利斯顿氏菌recA基因的特异性引物,并使用16S rRNA基因细菌通用引物扩增,引物如表1所示。16S rRNA基因和recA基因50μl扩增反应体系中含有:5 μl 10×buffer,MgCl22 mM,引物各0.2 μM,dNTPs各0.25 mM,Taq DNA聚合酶1.25 U,模板DNA约50 ng。反应在热循环仪上进行,16S rRNA扩增的循环参数为:94 ℃预变性10 min、94 ℃变性30 s、55 ℃复性45 s、72 ℃延伸90 s,35个循环后72 ℃保温10 min。recA基因扩增的循环参数为:94 ℃预变性10 min、94 ℃变性30 s、55 ℃复性45 s、72 ℃延伸40 s,35个循环后72 ℃保温10 min。反应完毕后分别取5 μl于1%琼脂糖凝胶电泳,凝胶成像系统下分析结果。PCR扩增产物送交上海生物工程技术公司测序。1.2.6 序列分析及数据处理 通过Blast软件从GenBank数据库中检索与16S rRNA基因和recA基因同源性较高的细菌基因序列,并从中选取与所待测细菌16S rRNA基因和recA基因同源序列,采用Clustal X 2.0软件[20]进行多序列匹配排列(Multiple Alignments),采用RDP3.34软件检测recA基因同源序列之间的基因重组事件。使用DNASTAR软件分别计算16S rRNA基因和recA基因的序列相似性。用系统发生推断软件包MEGA4.0[21]进行系统发育分析,计算recA基因的GC含量和碱基置换和颠换的比例;在Kimura-2-parameter 模型的基础上,用Neighbor-joining 法分别构建分子系统树,自举分析(Bootstrap)1000次重复检测分子系统树的置信度,缺失和不确定的位点在计算中被省略,使用其他的建树方法构建进化树比较确定NJ树聚类关系的准确性。

表1 PCR扩增的引物Tab.1 the primers used in the PCR amplifications
2 结 果
2.1 细菌分离
从上述患病香鱼的肝胰脏和肾脏中分离得到的细菌,在ZOBELL 2216E和TCBS培养基平板上分别得到形态特征相似的菌落。经过3次的平板划线纯化得到11株细菌其编号分别为:X0901至X0911。将纯化后的细菌接种于ZOBELL 2216E海水培养基试管斜面,30℃培养18 h后,于4℃冰箱中保存供鉴定和基因组DNA提取使用。
2.2 人工感染试验
自然发病的香鱼体表充血溃烂、肛门红肿、腹部肿胀等。香鱼在接种菌悬液后12 h内全部发病死亡;对照组的10尾香鱼养殖观察10 d均正常存活。剖检可见接种菌悬液香鱼临床特征与自然病例样类似但症状较轻。以菌液感染死亡的香鱼肝胰脏和肾脏为材料,通过平板分离纯化的细菌与供试菌类似,对健康的香鱼进行重复感染,同样可得到与自然病例样类似但临床症状较轻的病变。供试菌为香鱼出血溃烂病的病原菌。
2.3 形态及菌落特征
2.4 生理生化特征
对致原菌作生化管鉴定,内容和结果见表1。11株细菌的生理生化特征完全相同,均为氧化酶阳性、接触酶阳性、甘露醇阳性、吲哚阳性、硝酸盐还原阳性、MR测定阳性、精氨酸脱羧酶阳性、精氨酸双水解酶阳性;VP测定为阴性;利用葡萄糖、甘露糖和蔗糖产酸,参考《伯杰鉴定细菌学手册》(第9版)[22]得知与鳗利斯顿氏菌的生理生化特征非常相似。

表2 分离菌株的生理生化特征Tab.2 Biochemical and physiological characteristics of Strains
2.5 细菌DNA提取结果及扩增
细菌基因组DNA的纯度和完整性较好适合作PCR扩增模板。16S rRNA基因和recA基因的电泳结果如图1所示。11株致病菌的16S rRNA基因和recA基因序列测定结果完全相同,表明11株致病菌属于同种细菌,序列已提交到GeneBank数据库,登录号分别为:GQ4098612、GQ409861。
16S rRNA基因扩增片段的长度为1 393 bp,约占16S rRNA基因总长度的93%,扩增片段的GC含量为54.4%。用于建树的16S rRNA基因置换与颠换的比率k(嘌呤)= 1.719;k(嘧啶)= 3.084,置换颠换的总体偏差R=1.172。recA基因扩增片段长度为534 bp,约占recA基因总长度的50%,扩增片段的GC含量为45.3%。用于建树的recA基因置换与颠换的比率k(嘌呤)=2.646;k(嘧啶)= 3.796,置换颠换的总体偏差R=1.513。DNASTAR计算得出致病菌16S rRNA基因与鳗利斯顿氏菌16S rRNA基因的同源性大于99.9%;recA的同源性为99.8%。使用RDP3.34软件并没有检测到不同物种之间存在基因重组事件。利用16S rRNA基因和recA基因构建的进化树的结果如图2、图3所示。

图 1 16S rRNA基因和recA基因PCR扩增产物电泳结果

图 2 根据16 S rRNA基因序列构建的系统发育树Fig.2 Molecular phylogenetic tree based on 16 S rRNA gene sequences
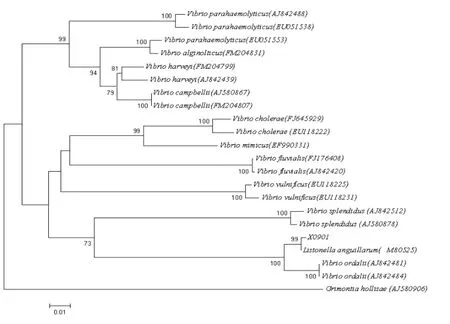
图 3 根据recA基因构建序列构建的系统发育树Fig.3 Molecular phylogenetic tree based on recA gene sequences
3 讨 论
本实验从出血溃烂病香鱼体内分离出11株致病菌,通过对致病菌的生理生化特征鉴定表明致病菌为鳗利斯顿氏菌。在细菌的系统发育地位研究方面,16S rRNA基因是最主要的分子工具,被广泛的应用于各种微生物系统发育地位研究。细菌分类学家普遍认为16S rRNA的同源性大于97.5%的细菌可视为同种。
随着新物种的发现,许多不同物种之间16S rRNA的同源性远大于97.5%,已不能够准确反映它们发育学地位。在这种前提下,国内外学者纷纷将视线转移到其他靶位点。Zeigler[16]提出用于系统发育研究的靶基因应该满足4个标准,即:1.靶基因必须广泛的分布于所有生物的基因组中;2.靶基因必须在给定物种的基因组中只出现一次;3.靶基因序列必须足够长,包含足够的信息,以及便于测序研究;4.序列必须能够预测所有物种的亲缘关系,并具有可以接受的精度和准确度,聚类结果与16S rRNA的鉴定结果和DNA
分子杂交结果相符。同时指出单个基因也可以很好的预测整个基因组的系统发育关系。recA基因是广泛存在于细菌中重要的持家基因,编码一种多功能蛋白质[17,18]。recA基因被用于研究Burkholderia属所有细菌的分类,证明是有效的鉴定工具[19]。Thompson等[14]利用recA做标记研究弧菌科细菌的系统发育关系,表明recA基因比16S rRNA具有更好的辨别能力,适合做为弧菌科细菌鉴定的工具。本研究为进一步确定致病菌的系统发育学地位,我们使用细菌16S rRNA通用引物并设计鳗利斯顿氏菌recA基因的特异性引物,扩增了致病菌的16S rRNA基因和recA基因的部分片段,并分别构建系统发育树。建树结果表明致病菌均能够和鳗利斯顿氏菌聚为一类,16S rRNA基因和recA基因的序列同源性分别为99.9%和99.8%,聚类结果与生理生化鉴定结果相一致。比较两株进化树可以得知相同弧菌在进化树上的拓扑学位置大致相符合。
Listonella anguillarum与Vibrio ordalii具有极近的亲缘关系,16S rRNA基因和recA基因的序列同源性分别为99.0%、97.6%,这一结果和DNA杂交试验得出的结论相一致[23];Vibrio cholerae与Vibrio mimicus亲缘关系较近,但是比较两株进化树可知它们的发育学地位并不完全一致,根据16S rRNA基因构树结果两种不同的细菌混杂在一起,而依据recA基因建树的结果能够把两种不同的细菌分开,Vibrio cholerae与Vibrio mimicus的16S rRNA基因和recA基因的序列同源性分别为99.4% ~ 99.6%、92.3% ~ 93.1%。Vibrio
parahaemolyticus、Vibrio alginolyticus、Vibrio harveyi和Vibrio campbellii四种细菌的亲缘关系相对较近,建树结果均可以将它们聚为一枝,但是Vibrio harveyi在16S rRNA构建的进化树中并没有彼此聚在一起,产生这种结果的原因是由于这4株细菌的16S rRNA的序列彼此之间同源性大于99%,建树方法已无法对它们分类学地位作出准确的估计。在以recA基因构建的进化树中Vibrio parahaemolyticus(EU051553)和Vibrio alginolyticus聚在一起,产生这种结果的原因尚不明确,需要做进一步研究确定这两种菌的分类地位。Vibrio fluvialis和Vibrio vulnificus的拓扑学结构并不相符,产生这种情况可能的原因有两种,一是“长枝吸引效应”,是指在用系统发育分析方法分析一个有限数据集时, 由于高频率的相似变化(如趋同、平行进化)和加速的进化速率等因素的存在使序列达到相同状态而人为地将这些不是来自于共同祖先的序列的代表分类元聚在一起, 使这些分类元之间相互“吸引”[24]。在使用外围集团确定树根的过程中,一个明显距离很远的外围集团很可能会拥有一个分歧非常大的序列,以至于内部集团将受到长树枝效应的影响,把这个序列同内部集团放在一起[25]。二是recA片段提供的进化信息不足,以致出现分类学地位模糊的现象。对上述结果产生的原因还需要再做进一步的研究。总体来说:与16S rRNA基因相比,recA基因在弧菌科常见细菌的系统发育地位分析中具有更好的鉴别能力。
[1]国家环保局, 中华人民共和国濒危物种科学委员会.中国濒危动物红皮书(鱼类)[M].北京: 科学出版社, 1998, 47-49.
[2]叶岩豹.香鱼增养殖技术 [M].杭州: 浙江科学技术出版社, 2004.
[3]Bonaveri G F.Quoted in Drouln de Bouville R.de Les maladies des poissons d'eau douce d'Europe [J].Annal Sc.Agronom, 1907, 1: 120-250.
[4]Bergman A M.Die rote Beulenkrankheit des Aals Ber Kgl Bayer [J].Biol Verssta, 1909, 2: 10-54.
[5]MacDonell M T, Colwell R R.Phylogeny of theVibrionaceae, and recommendation of two new genera,ListonellaandShewanella[J].Syst Appl Microbiol, 1985, 6: 171-182.
[6]Toranzo A E, Santos Y, Lemos M L.Homology ofVibrioan anguillarumstrains causing epizooticsin turbot salmon and trout reared on the Atlantic coast of Spain [J].Aquaculture, 1987, (67): 41-52.
[7]肖慧, 李军, 王祥红, 等.鲈鱼苗烂鳃、烂尾病病原菌的研究 [J].青岛海洋大学学报, 1999, 29(1): 87-93.
[8]E Myhr, J L Larsen, A Lillehaug, et al.Characterization ofVibrio anguillarumand closely related species isolated from farmed fish in Norway [J].Appl Environ Microbiol, 1991 September; 57(9): 2 750-2 757.
[9]莫照兰, 茅云翔, 陈师勇, 等.一株牙鲆皮肤溃烂症病原菌的鉴定 [J].微生物学报, 2002, 42(3): 263-265.
[10]张晓君, 秦国民, 陈翠珍, 等.大菱鲆病原鳗利斯顿氏菌的药物敏感性测定与分析 [J].海洋通报, 2008, 27(5): 35-38.
[11]李清禄, 陈强.海水网箱养殖大黄鱼细菌性病原鉴定与感染治疗研究 [J].应用于环境生物学报, 2001, 7(5): 489-493.
[12]塞文婴, 东秀珠.定向进化同源基因在细菌系统进化研究中的应用 [J].微生物学通报, 2000, 27(5): 377-381.
[13]李宁求, 白俊杰, 吴淑勤, 等.斜带石斑鱼3种致病性弧菌的分子生物学鉴定 [J].水产学报, 2005, 20(3): 356-361.
[14]Thompson C C, Thompson K, Vandemeulebroecke K, et al. Use of recA as an alternative phylogenetic marker in the family Vibrionaceae [J].International Journal of Systematic and Evolutionary Microbiology.2004, 54, 919-924.
[15]Thompson F L, Gevers D, Thompson C C, et al.Phylogeny and Molecular Identification of Vibrios on the Basis of Multilocus Sequence Analysis [J].Applied And Environmental Microbiology.Sept.2005, 71(9): 5 107-5 115.
[16]Zeigler D R.Gene sequences useful for predicting relatedness of whole genomes in bacteria [J].Int J Syst Evol Microbiol.2003, 53, 1 893-1 900.
[17]Cox M M.The bacterial recA protein as a motor protein [J].Annu Rev Microbiol.2003, 57: 551-577.
[18]Lloyd A T, Sharp P M.Evolution of the recA gene and the molecular phylogeny of bacteria [J].J Mol Evol.1993, 37, 399-407.
[19]Mahenthiralingam E, J Bischof, S K Byrne, et al.DNA-Based diagnostic approaches for identification ofBurkholderia cepacia complex,Burkholderia vietnamiensis,Burkholderia multivorans,Burkholderia stabilis, andBurkholderia cepaciagenomovars I and III [J].J Clin Microbiol, 2000, 38: 3 165-3 173.
[20]Thompson J D, Gibson T J, Plewniak F, et al.The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools [J].Nucleic Acids Research, 1997, 25: 4 876-4 882.
[21]Tamura K, Dudley J, Nei M, et al.MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0 [J].Molecular Biology and Evolution.2007, 24: 1 596-1 599.
[22]Holt J G, Krieg N G, Sneath P H A, et al.Bergey’s manual of determinative bacteriology, 9th Edition [M].Baltimore: Williams and Wilkins, 1994.382-390.
[23]J J Farmer.The family Vibrionaceae [M].Prokaryotes.2006.6: 495-507.
[24]黎一苇, 于黎, 张亚平.系统发育研究中“长枝吸引”假象概述 [J].遗传, 2007, 29(6): 659-667.
[25]Andreas D, Baxevanis B F.Francis Ouellette.生物信息学 基因和蛋白质分析的实用指南 [M].清华大学出版社, 2000.1: 176-208.
Isolation and identification ofListonella anguillarumfromPlecoglossus altivelisbased on 16S rRNA and recA
WANG Cheng-yi1,2,3,YAN Mao-cang2,3,CHEN Shao-bo2,3,GUAN Min-xin1,4,SHAN Le-zhou2,3,AI Wei-ming1,XIE Qi-lang2,3,CAI Yan-ben5
(1.Wenzhou Medical College, 325005, China; 2.Zhejiang Mariculture Research Institute, 325005, China; 3.Zhejiang Key Laboratory of Exploitation and Preservation of Coastal Bio-resource, 325005, China; 4.Division of Human Genetics and Center for Hearing and Deafness Research, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio 45229, USA; 5.Wenzhou Engineering Graduate School, 325028, China)
In order to investigate the pathogen of bleeding ulcers ofPlecoglossus altivelisand the phylogenetic position of this pathogen, 11pathogen were isolated from liver, kidney and surface of diseasedPlecoglossus altiveliswith the NO.from X0901 to X0911.Routine bacterial identification was used to identify this pathogen and the phylogenetic position was investigated using 16S rRNA gene and recA gene sequences analysis.The results of physiological and biochemical tests suggested that they were gram-negative bacteria, and the results of oxidase, contact enzyme, producing indole, arginine decarboxylase, arginine dihydrolase, nitrate reduction tests and MR tests were positive; but they couldn’t generate H2S and the results of V-P test were negative too.Molecular phylogenetic trees of common bacterial were constructed and the results suggested that the pathogen andListonella anguillarumclustered with each other.And the results corresponded with that of routine bacterial identification.The identity of 16S rRNAgene and recA gene between pathogen andListonella anguillarumwere 99.9% and 99.8% respectively.The topology of phylogenetic tree constructed by recA gene was generally agreed with that of 16S rRNA gene.And recA gene was much more discriminatory than 16S rRNA gene.So the conclusion was recA gene was an alternative phylogenetic and identification marker for the common vibrios.
Plecoglossus altivelis; Listonella anguillarum; isolation; identification
P736.22+1
A
1001-6932(2010)01-0084-07
2009-09-22;
2009-11-06
浙江省科技厅科研院所专项公益技术攻关项目 (2007F10009);浙江省科技厅科研院所青年人才计划项目 (2006R20001);温州市科技兴海项目 (S20070051)
王成义 (1984-),男,在读硕士,主要病害及遗传学研究。电子邮箱:sciencew@163.com
陈少波, chenshaobo@hotmail.com
