CnAl+小团簇结构的几何特征与稳定性
2010-12-05马文瑾张献明武海顺
马文瑾 宋 翔 张献明 武海顺
(山西师范大学化学与材料科学学院,山西临汾 041004)
CnAl+小团簇结构的几何特征与稳定性
马文瑾*宋 翔 张献明 武海顺*
(山西师范大学化学与材料科学学院,山西临汾 041004)
采用密度泛函理论(DFT)的B3LYP方法,研究了CnAl+(n=2-12)团簇的几何结构与电子性质.在6-311++ G**水平上对CnAl+(n=2-12)团簇进行了几何构型优化和振动频率计算.结果表明,CnAl+团簇的基态结构为Al原子与Cn链端基配位形成的直线或折线形结构,以及Al原子与Cn环上1个C原子端位相连或打开Cn环与2个C原子相连形成的环状结构.分子总的平均键长随着n的增大逐渐趋于定值(0.138 nm).通过对基态结构的能量分析,得到了CnAl+团簇的稳定性信息.
密度泛函理论;CnAl+团簇;基态结构;稳定性
近年来,含杂原子碳团簇的理论和实验研究已有报道[1].用激光溅射固体样品实验可观察到一系列含杂原子的碳团簇[2-5].由于掺入铝原子使得碳团簇的结构和电子特性发生了明显的变化.因此,人们期望从理论上进一步揭示各种富碳掺铝团簇的形成机理.随着CnAlm团簇理论和实验研究的不断深入[6-21],对CnAl±团簇的理论与实验研究正引起了人们的关注[4,5,22-24].Liu等[4-5]通过飞行时间质谱实验和从头算的Hartree-Fock(HF)方法分别得到了团簇的飞行时间质谱图和几何结构与稳定性规律.李光平等[22]采用HF和单、双激发组态相互作用(CISD)方法研究了AlCn和(n=1-4)团簇的几何结构、原子化能以及原子平均结合能等.Boldyrev等[23]分别采用B3LYP、耦合簇CCSD(T)和外壳层格林函数(OVGF)方法研究了团簇的几何结构和振动频率与光电谱图.Largo等[24]采用B3LYP方法研究了AlCn、(n=1-7)团簇的几何结构和振动频率与偶极矩.然而对CnAl+(n>7)团簇的理论研究至今尚未见报道.本文对CnAl+(n=2-12)团簇的几何结构和稳定性规律进行了理论研究,其结论对理解小尺寸团簇的形成机理以及寻找更大尺寸团簇的理论研究可提供有意义的参考.
1 计算方法
首先采用穷举法对CnAl+(n=2-12)团簇的各种可能构型进行了结构设计和点群确定.理论优化过程先是在HF/6-31G*水平上对所有可能构型考虑不同自旋多重态进行初次结构优化,并用振动频率验证计算构型存在的可能性;其次是将初次优化得到的各团簇中能量较低的结构及可能存在的对称性进行点群调整,作为初始参数重新在B3LYP/6-31G*水平上进行结构优化,从而确定能量较低的稳定结构;最后对第二步优化所得能量较低的稳定结构在更高层次上用B3LYP/6-311++G**方法进行更为精确的优化和频率计算,最终确定了CnAl+(n=2-12)团簇的基态结构.全部工作均采用Gaussian 03程序[25],在山西师范大学材料化学研究所完成.
2 结果与讨论
2.1 基态结构的几何构型
由B3LYP/6-311++G**理论优化得到的CnAl+(n=2-12)团簇基态结构中的所有原子均处在同一平面内.n≤9时,n为奇数的CnAl+团簇均为Al原子处于Cn链端位的线状结构;n为偶数的CnAl+团簇分别为Al原子与Cn链端位相连的折线形结构或Al原子中心插入Cn环上1个C—C键中的环状结构;n>9时均为Al原子与Cn环上1个C原子端配位的环状结构.为了节省篇幅,图1只给出了各线状和环状基态结构的示意图,其中线状结构的原子编号自左至右顺序为1、2、3、…等;环状结构的原子编号自Al原子起由左至右并按顺时针方向依次为1、2、3、…等.表1为这些团簇基态结构的对称性和几何参数,括号中为相应的电子态.
n=3,5,7,9奇数结构中的Al原子总是与Cn链端位1个C原子相连形成分子点群为C∞v和近似于C∞v对称性的Cs线状结构.与文献[22]和[24]报道的基态结构相一致.结构中的C—Al键长变化范围为0.218-0.254 nm.C—C键长的变化范围为0.124-0.133 nm,介于叁键(0.120 nm)和双键(0.134 nm)之间[10],且Cn链具有叁键和双键交替变化的特征.n=3,5,7结构中的C—Al和C—C平均键长分别为0.237和0.128 nm,与文献[24]报道的相应平均键长0.237和0.128 nm完全一致.具有Cs对称性的C9Al+结构中,Al(1)—C(2)—C(3)键角为179.9°,几乎为平角.
n=2,4,6,8偶数的平面结构中,n=2,6,8时分别为Al原子与C—C键边桥配位或打开Cn环上1个C—C键中心插入与2个C原子相连形成分子点群为C2v对称性的环状平面结构;n=4时为Al原子与Cn链端基配位形成具有Cs对称性的折线状平面结构.结构中的C—Al键长变化范围为0.184-0.203 nm.C—C键长的变化范围为0.124-0.137 nm,介于叁键和单键(0.154 nm)之间[10],形成类似于C≡C—C≡C单键和三键交替的聚乙炔化合物结构特征.环状的C2Al+和C6Al+结构分别比文献[24]报道的直线和折线状结构能量低0.84和0.27 eV(1 a.u.= 27.212 eV),其中C2Al结构的C—Al平均键长为0.203 nm,比文献[22]报道的环状基态结构C—Al平均键长0.202 nm仅伸长了0.001 nm;C—C键长0.127 nm,与文献[22]报道的键长0.127 nm完全一致.折线状C4Al+团簇比文献[22]报道的直线形结构能量低0.12 eV.结构中C(3)—C(4)—C(5)键角为161.5°,与文献[24]报道的C—C—C键角170.7°相差9.2°. C—Al键长0.184 nm,与文献[22]和[24]报道的键长0.185和0.184 nm相比可以看出,前者仅缩短了0.001 nm,后者则完全一致;C—C平均键长0.128 nm,比文献[22]报道的平均键长0.127 nm伸长了0.001 nm,与文献[24]报道的平均键长0.128 nm具有很好的一致性.

图1 CnAl+团簇的几何构型Fig.1 Geometric configuration of CnAl+clusters

表1 CnAl+基态结构的对称性和几何参数Table 1 Symmetry and geometric parameters of the ground state structures of CnAl+
n=10,11,12结构中的Al原子倾向于与Cn环上1个C原子端位相连形成分子点群分别为C2v和Cs对称性的环状平面结构.结构中的C—Al键长随着n的增大逐渐缩短,3个C—Al键长分别为0.229、0.223和0.221 nm.Cn环中C—C键长的变化范围为0.123-0.143 nm,介于叁键和单键之间,平均键长略有伸长.分子总的平均键长趋于定值0.138 nm.
由以上构型分析可以看出,CnAl+(n=2-12)团簇基态结构有两种基本构型,n≤9时为Al原子与Cn链端位相连形成的直线或折线状结构,以及Al原子与C—C键边配位或打开Cn环上1个C—C键中心插入与2个C原子相连形成的环状结构;n>9时,均为Al原子与Cn环上1个C原子端配位的环状结构.随着n的增大,n≤9时的C—Al和C—C平均键长均呈现奇偶长短交替变化的特征;n>9时C—Al键长逐渐缩短, C—C平均键长略有伸长.分子总的平均键长随着n的增大逐渐趋于定值0.138 nm.
在确定簇合物CnAl+(n=2-12)基态结构的过程中,分别计算了图1所示结构的振动频率,得到了CnAl+(n=2-12)基态结构的振动频率ν和振动强度I.表2只给出了每一基态结构最小振动频率和振动强度最大值对应的振动频率,括号中为对称振动方式. ν值最小的振动频率可以反映所得结构是否存在虚频,I值最大对应的振动频率可以反映红外光谱中最强吸收峰的位置.由表2可以看出,所有结构振动频率的波数均为正值,表明优化所得结构均为势能面上的稳定点.
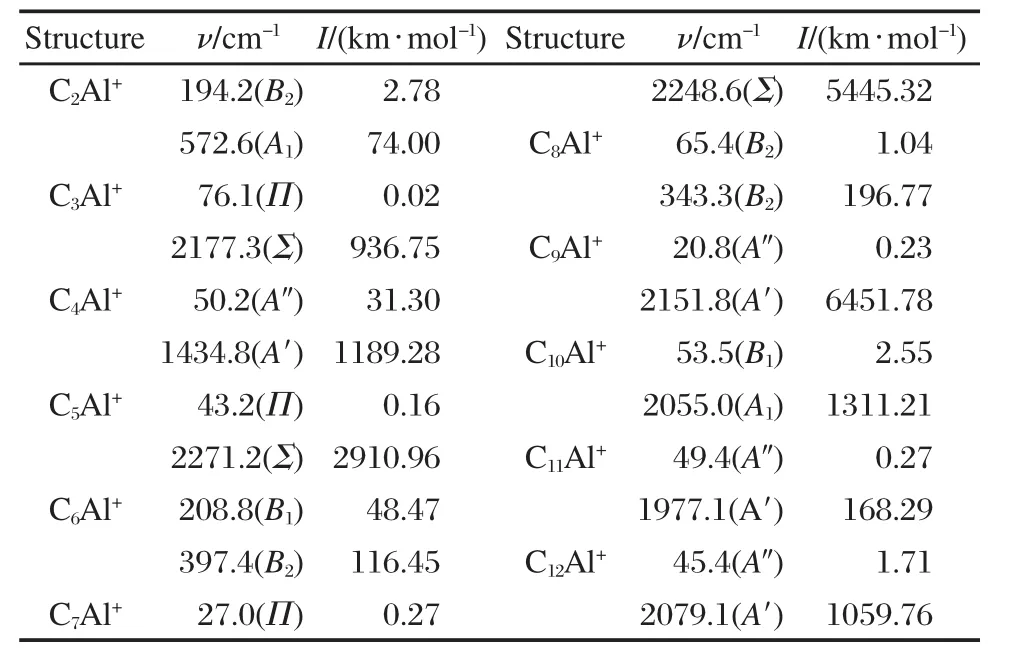
表2 CnAl+团簇基态结构的振动频率Table 2 Vibrational frequencies of the ground state structures of CnAl+clusters
由自然键轨道(NBO)得到文献[19]中CnAl团簇基态结构各原子上的净电荷分布可以看出,在Al和Cn相互作用形成CnAl团簇基态结构的过程中,Al原子上电荷发生了向C原子上的转移,这种电荷转移的作用使得C—Al端键上的C原子呈负电性,Al原子显正电性,C—Al键离子化.分析CnAl到CnAl+结构的电荷分布可知,电离1个电子的CnAl+结构中,Al原子得电子倾向比相应的中性结构明显增大, C原子电负性略有降低.Cn链上各原子由于C—C成键轨道上净电荷分布存在差异,导致C—C键长出现长短交替变化的特征[26].
2.2 基态结构的稳定性
2.2.1 原子化能和热力学性质
为了寻求CnAl+(n=2-12)团簇基态结构随n变化的规律,表3给出了各团簇基态结构的总能量(ET)、零点能(Ez)、摩尔热容(Cp)和标准熵(S⊖).可以看出,Ez,Cp和S⊖数值随着n的增大呈现增大趋势,其中Ez数值近似线性增大,平均增大幅度约为14.03 kJ·mol-1.
在研究CnAl+团簇稳定性规律的过程中,考虑了如下化学反应:

相应的能量变化定义为


表3 CnAl+团簇基态结构的能量及热力学性质Table 3 Total energies and thermodynamical parameters of the ground state structures of CnAl+clusters
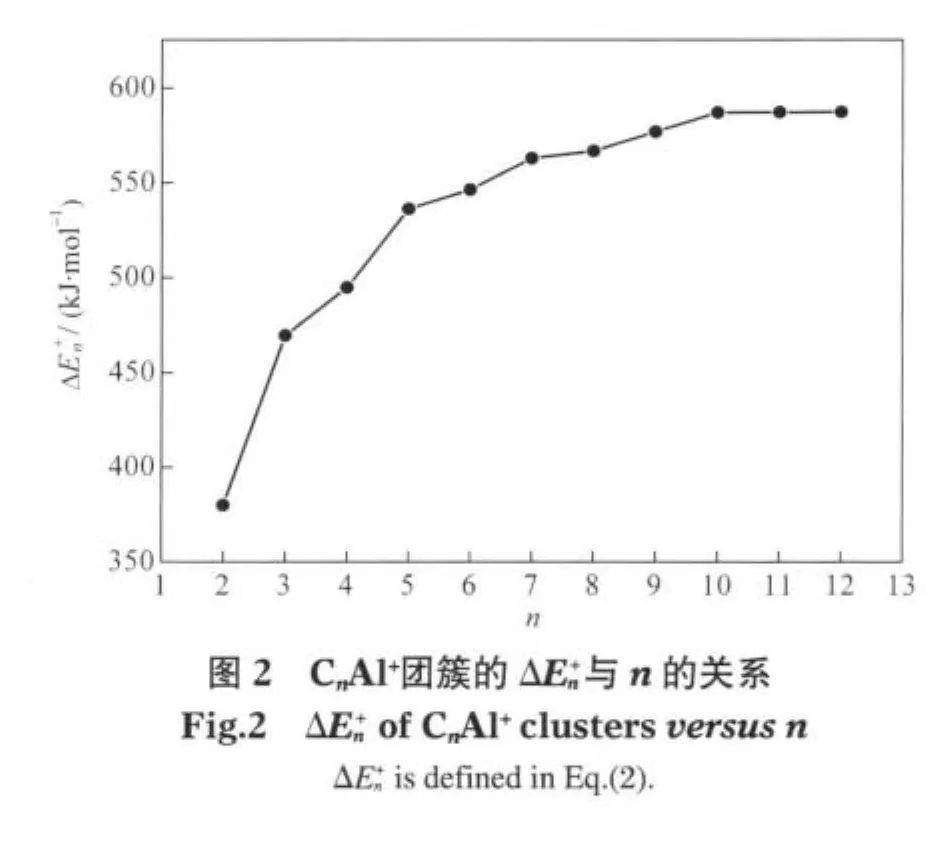
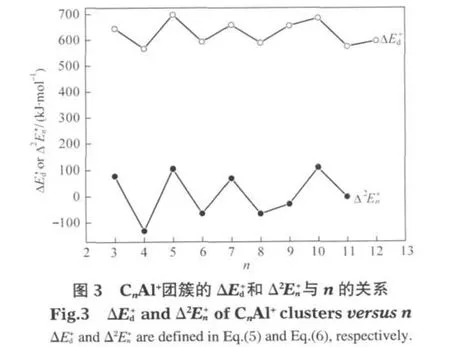
2.2.2 离解能和能量二次差分值
为了进一步考察CnAl+团簇的稳定性,还考虑了如下两类化学反应:

相应的能量变化分别定义为
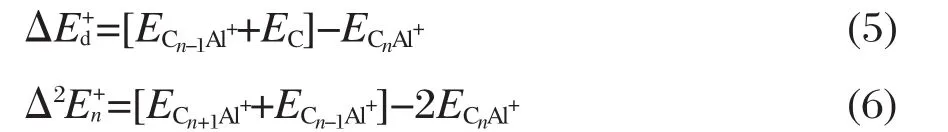
3 结论
CnAl+(n=2-12)团簇基态结构有线状和环状两种基本构型,结构中的所有原子均处在同一平面内. n≤9时,n为奇数的CnAl+团簇均为Al原子与Cn链端位相连的线状结构;n为偶数的CnAl+团簇分别为Al原子处于Cn链端位的折线形结构和Al原子与C—C键边桥配位或打开Cn环与2个C原子相连的环状结构.n>9时均为Al原子与Cn环上1个C原子端基配位的环状结构.随着n的增大,n≤9时的C—Al和C—C平均键长均呈现奇偶长短交替变化的特征;n>9时C—Al键长逐渐缩短,C—C平均键长略有伸长.分子总的平均键长逐渐趋于定值0.138 nm.n≤9的CnAl+团簇基态结构中,n为奇数的稳定性好;n>9的CnAl+团簇基态结构中,n为偶数时较稳定.
1 Liu,J.W.;Chen,M.D.;Zheng,L.S.J.Phys.Chem.A,2004,108: 5704
2 Becker,S.;Dietze,H.J.Int.J.Mass Spectrom.Ion Process.,1988, 82:287
3 Consalvo,D.;Mele,A.;Stranges,D.;Giardini,G.A.;Teghil,R. Int.J.Mass Spectrom.Ion Process.,1989,91:319
4 Liu,Z.Y.;Wang,C.R.;Huang,R.B.;Zheng,L.S.Int.J.Mass Spectrom.Ion.Process.,1995,141:201
5 Liu,Z.Y.;Huang,R.B.;Tang,Z.C.;Zheng,L.S.Chem.Phys., 1998,229:335
6 Bauschlicher,J.C.W.;Langhoff,S.R.;Pettersson,L.G.M. J.Chem.Phys.,1988,89:5747
7 Brazier,C.R.J.Chem.Phys.,1993,98:2790
8 Chertihin,G.V.;Andrews,L.;Taylod,P.R.J.Am.Chem.Soc., 1994,116:3513
9 Yang,H.L.;Tanaka,K.;Shinada,M.J.Mol.Struct.-Theochem, 1998,422:159
10 Zheng,X.E.;Wang,Z.Z.;Tang,A.C.J.Phys.Chem.A,1999, 103:9275
11 Li,X.;Wang,L.S.;Boldyrev,A.I.;Simons,J.J.Am.Chem.Soc., 1999,121:6033
12 Cannon,N.A.;Boldyrev,A.I.;Li,X.;Wang,L.S.J.Chem.Phys., 2000,113:2671
13 Barrientos,C.;Redondo,P.;Largo,A.Chem.Phys.Lett.,2000, 320:481
14 Ashman,C.;Khanna,S.N.;Pederson,M.R.Chem.Phys.Lett., 2000,324:137
15 Kawamata,H.;Negishi,Y.;Nakajima,A.;Kaya,K.Chem.Phys. Lett.,2001,337:255
16 Zhao,J.J.;Liu,B.C.;Zhai,H.J.;Zhou,R.F.;Ni,G.Q.;Xu,Z.Z. Solid State Commun.,2002,122:543
17 Midda,S.;Das,A.K.J.Mol.Struct.-Theochem,2003,633:67
18 Largo,A.;Redondo,P.;Barrientos,C.J.Am.Chem.Soc.,2004, 126:14611
19 Wang,Y.B.;Ma,W.J.;Zhang,J.;Wu,H.S.Acta Phys.-Chim. Sin.,2007,23:873 [王艳宾,马文瑾,张 静,武海顺.物理化学学报,2007,23:873]
20 Ma,W.J.;Zhang,X.M.;Xu,X.H.;Wang,Y.B.;Wu,H.S.Acta Phys.-Chim.Sin.,2008,24:1477 [马文瑾,张献明,许小红,王艳宾,武海顺.物理化学学报,2008,24:1477]
21 Naumkin,F.Y.J.Phys.Chem.A,2008,112:4660
22 Li,G.P.;Zhang,H.B.;Tian,A.M.;Yan,G.S.Acta Phys.-Chim. Sin.,1995,11(3):211 [李光平,张华北,田安民,鄢国森.物理化学学报,1995,11(3):211]
23 Boldyrev,A.I.;Simons,J.;Li,X.;Wang,L.S.J.Am.Chem.Soc., 1999,121:10193
24 Largo,A.;Redondo,P.;Barrientos,C.J.Phys.Chem.A,2002, 106:4217
25 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision B.04.Pittsburgh,PA:Gaussian Inc.,2003
26 Fan,Q.;Gary,V.;Pfeiffer,G.V.Chem.Phys.Lett.,1989,162: 472
27 Bonacic,K.V.;Fantucci,P.;Koutecky,J.Chem.Rev.,1991,91: 1035
November 2,2009;Revised:January 7,2010;Published on Web:March 30,2010.
Structural Characteristics and Stability of CnAl+Small-Size Clusters
MA Wen-Jin*SONG Xiang ZHANG Xian-Ming WU Hai-Shun*
(School of Chemistry and Material Science,Shanxi Normal University,Linfen 041004,Shanxi Province,P.R.China)
The geometric and electronic properties of CnAl+(n=2-12)clusters were investigated using the B3LYP method of density functional theory(DFT).Structural optimization and frequency analyses were performed with the 6-311++G**basis set.Calculation results showed that the ground state of the CnAl+clusters was a linear or polyline structure with a terminal aluminum atom,and an aluminum atom was inserted into the Cnring to form a new ring structure or an aluminum atom bonded to one side of the monocyclic Cnring.With an increase in n,the total average molecular bond length gradually approached 0.138 nm.We obtained stability information by an energy analysis of the ground state.
Density functional theory; CnAl+clusters; Ground state structure; Stability
[Article] www.whxb.pku.edu.cn
*Corresponding authors.Email:ma_w_j@163.com,wuhs@dns.sxtu.edu.cn;Tel:+86-35-72052468.
The project was supported by the National Natural Science Foundation of China(20771069,20871077)and University Science and Technology Projects of Shanxi Province,China(20091015).
国家自然科学基金(20771069,20871077)和山西省高校科技项目(20091015)资助
O641
