PW11/SBA-15介孔杂化材料的直接合成与表征
2010-11-30王金娥
王金娥 杨 春
(南京师范大学化学与环境科学学院,江苏省生物功能材料重点实验室,南京 210097)
PW11/SBA-15介孔杂化材料的直接合成与表征
王金娥 杨 春*
(南京师范大学化学与环境科学学院,江苏省生物功能材料重点实验室,南京 210097)
采用三嵌段共聚物EO20PO70EO20(P123)为模板剂,正硅酸乙酯(TEOS)和缺位Keggin型多金属氧酸盐Na7PW11O39(PW11)为无机前驱体,由共缩合法一步合成了PW11/SBA-15介孔杂化材料.通过红外(IR)光谱、紫外-可见漫反射光谱(UV-Vis/DRS)、X射线衍射(XRD)、低温N2吸附、透射电子显微镜(TEM)等手段对杂化材料和合成过程进行了表征.结果表明:杂化材料中不仅多金属氧酸盐的Keggin单元保留完整,且共价键联于介孔孔道内部,而且样品基本具有规整有序的六方介孔结构.TEOS预水解时间的长短对有序结构的构筑有明显影响,随预水解时间延长,样品的介观有序性增加.这是因为多金属氧酸盐对模板剂P123有盐析作用,其作用大小与多金属氧酸盐前驱物的溶解度有关.
介孔分子筛;多金属氧酸盐;杂化材料;Na7PW11O39;SBA-15
由于具有纳米尺寸的规整孔道结构、高的比表面积和热稳定性,介孔分子筛材料在吸附、催化和纳米材料的组装、制备方面有广泛应用.利用介孔材料的宽敞孔道空间,通过表面功能化的方法将一些体积较大的功能分子和基团修饰、组装到孔道内部,制备性能优良的吸附剂、催化剂和其它功能材料已成为近十年来该领域的研究热点[1-6].多金属氧酸盐(POM)是一类功能优异的无机簇状配合物,在催化、医药、光、电、磁以及燃料电池等领域都有应用.自介孔分子筛问世以来,人们便开始探索将POM与具有有序孔结构的载体材料相结合,以获得表面积大、稳定性高、并兼具POM功能的新型功能材料.物理吸附于介孔载体上的负载型POM催化剂已有大量研究[7-11],其明显的缺点在于POM不能牢固地固定在载体上,容易被极性溶剂浸取而溶脱流失.因此,近年来人们开始探索用化学方法将其固定在载体表面[12-17].
我们根据介孔分子筛表面功能化的基本原理和途径,提出了将POM共价键联于介孔载体表面的新思路,并采用共缩合法和二次嫁接法分别成功合成了SiW11/SBA-15和PW11/SBA-15介孔杂化材料[18-20].但对二次嫁接得到的PW11/SBA-15,在键联负载量较高的情况下,POM在孔内分布不均,孔道出现了部分堵塞的现象[20].用共缩合法直接合成的SiW11/SBA-15则不存在这样的问题.因此本文采用与SiW11/SBA-15合成相似的方法,以Keggin型一缺位钨磷酸盐PW11和正硅酸乙酯(TEOS)为无机前躯体,三嵌段共聚物EO20PO70EO20(P123)为结构导向剂,合成了具有六方结构的PW11/SBA-15介孔分子筛杂化材料,并与SiW11/SBA-15的合成进行了比较和讨论.
1 实 验
1.1 样品制备
Na7PW11O39(PW11)的合成根据文献[21-22]进行. (Bu4N)3PW11O39[O(SiOH)2]((Bu4N)3PW11Si2)的合成根据文献[23]进行.其中,有机硅烷用TEOS代替.具体步骤和产物表征见文献[20].
纯硅介孔分子筛SBA-15的合成根据文献[24-25]进行.介孔杂化材料PW11/SBA-15的合成与此相似,具体步骤如下:将1 g三嵌段共聚物P123 (Aldrich,Mav=5800)溶解于30 mL 2 mol·L-1盐酸中,升温至40℃.在此温度下滴加2.03 g(9.75 mmol) TEOS,并使其预水解一定时间.然后加入7.5 mL PW11水溶液(0.71 g(0.25 mmol)PW11溶于7.5 mL水).体系中各物质的摩尔比为n(TEOS)∶n(PW11)∶n(HCl)∶n(H2O)=0.00975∶0.00025∶0.06∶1.96.继续搅拌晶化24 h,再于80℃恒温箱中静置老化24 h.样品冷至室温后过滤,充分水洗,空气中晾干.所有介孔样品均采用抽提法脱除模板剂P123,即以乙醇为溶剂,用Soxhlet抽提器抽提样品72 h.
除特别说明外,上述合成中所用试剂均为国产市售分析纯试剂,使用时未进行进一步处理.实验用水为实验室用蒸馏水,元素分析(ICP-AES)用水为18.2 MΩ·cm超纯水.
1.2 样品表征
IR光谱测量用Bruker Tensor-27型(德国Bruker公司)FT-IR光谱仪进行,样品与KBr混合压片,扫描范围为4000-400 cm-1,仪器分辨率4 cm-1.UVVis/DRS(紫外-可见漫反射光谱)测定采用Varian Cary 5000型(美国Varian公司)紫外-可见-近红外分光光度计进行,扫描范围为 200-800 nm,固体BaSO4作参比.小角XRD分析在ARL X′TRA型(瑞士ARL公司)X射线衍射仪上进行,Cu Kα射线,石墨单色器,管电压40 kV,管电流40 mA,扫描范围2θ为 0.5°-6°.N2吸附-脱附数据在Micromertics ASAP 2020 M型(美国Micromertics公司)吸附仪上获得,样品120℃预脱气6 h.由BET法计算样品的比表面积;由BJH模型根据吸附等温线计算孔径分布.孔容为p/p0=0.98时的单点总孔容.TEM影像在JEOL JEM-2100型(日本JEOL公司)透射电子显微镜上测得,加速电压200 kV.电感耦合等离子体原子发射光谱(ICP-AES)在Leeman Lab Prodigy型(美国Leeman Lab公司)电感耦合等离子原子发射光谱仪上测得.
2 结果与讨论
用共缩合法合成共价键联的POM/SBA-15杂化材料的基本思路是:采用具有缺位Keggin结构的XW11(X=Si,P)作为POM前驱物,由于该结构中缺位处的表面氧原子具有很高的亲核性,能与硅酸酯中具有亲电性的Si原子反应,导致两个Si原子插入空位形成Si—O—W键,并使结构趋于饱和.这样,当XW11引入以TEOS为硅源进行的SBA-15的合成体系中时,TEOS不仅可自身缩合形成介孔氧化硅,而且可以同时与XW11反应,将其以共价键联于介孔孔道内部.这一思路我们已通过SiW11/SBA-15杂化材料的合成得以实现[18-19],PW11与TEOS间的反应也已被证实(我们用PW11与TEOS反应合成了具有饱和结构的PW11O39[O(SiOH)2]3-,即).因此通过这一途径合成PW11/SBA-15杂化材料是完全有可能的.另外,在SiW11/SBA-15的合成中我们发现,POM前驱物加入之前,须给TEOS自身的水解缩合以及孔壁构筑留有足够的时间(即预水解时间),否则,POM的盐析作用会导致模板剂P123析出,扰乱有序结构的组装和构造,导致合成失败.因此,在本文中我们将同样关注预水解时间对PW11/ SBA-15产物结构的影响.
图1示出了几个样品的IR谱图.与纯SBA-15 (图1a)相比,杂化样品PW11/SBA-15的IR谱(图1b)中除SBA-15本身的特征带外,在874 cm-1处多出了一个吸收带,约800 cm-1处的吸收带也移动至825 cm-1处.与(Bu4N)3PW11Si2的IR谱(图1d)对比可知,它们是结构中的W—O—W伸缩振动带,而结构中的其它谱带则被SBA-15自身的Si—O—Si不对称伸缩带(1080 cm-1)和O—Si—O不对称伸缩带(约960 cm-1)掩盖.图1b与图1a的差谱(图1c)则更清楚地显示出杂化材料中存在的结构,969、874、825、770、520 cm-1处的几个主要特征带都清楚地表现出来,说明在杂化样品中,不仅多金属氧酸盐的Keggin结构得以保留,而且PW11已与TEOS发生了反应,Si原子插入PW11的空位,形成了类似于PW11O39[O(SiO—)2]3-的结构.这样,多金属氧酸盐便以共价键键联于纯硅SBA-15的孔壁上了.元素分析表明,杂化样品中POM的含量(以PW11O39计)约为25.8%.
图2为样品的UV-Vis/DRS谱.由图可见,纯SBA-15(图2a)在紫外区无吸收峰;(Bu4N)3PW11Si2(图2b)在260-290 nm处有一宽的吸收峰;而杂化样品PW11/SBA-15(图2c)的谱图与(Bu4N)3PW11Si2的谱图相似,在约265 nm处有一强而锐的吸收峰,再一次表明在杂化材料的合成过程中,Keggin单元进入了SiO2骨架,且Si原子嵌插进入了PW11的空位,形成了近饱和的Keggin结构.杂化样品的吸收峰更窄,可能是其中多金属氧酸盐分散度更高的缘故.

图1 样品的IR谱Fig.1 IR spectra of samples(a)SBA-15,(b)PW11/SBA-15(prehydrolysis time=2 h),(c)subtracting(a)from(b),(d)(Bu4N)3PW11Si2

图2 样品的UV-Vis/DRS谱Fig.2 UV-Vis/DRS spectra of samples (a)SBA-15,(b)(Bu4N)3PW11Si2,(c)PW11/SBA-15(prehydrolysis time=2 h)
为了更好地了解样品合成过程中多金属氧酸盐各物种的变化情况,我们在PW11/SBA-15样品的合成过程中,对各个步骤的固体产物进行了取样分析,图3为其IR谱.由图3c可见,PW11一旦加入反应体系,一部分立即与TEOS作用形成了物种(约874 cm-1),另一部分则很快转变成了饱和结构的PW12(显示PW12的896 cm-1特征带),这些物种粘附、包夹在经预水解形成的SiO2中,并被过滤收集得到;对此SiO2固体进行水洗(图3d),物种和PW12的特征带均消失不见,表明此时生成的物种并没有与SiO2发生键联,与PW12一样均可水洗除去.40℃晶化后样品的IR谱(图3e)与图3c非常相似,但样品被充分水洗后,物种并未完全消失,有相当量的此物种被保留下来(图3f),表明它们已通过表面的SiOH与SiO2发生了键联. 80℃老化后(图3g),物种减少(与图3e相比),表明一些游离态的物种已在高温下水解,而键联的物种在水洗后仍然保留在样品中(图3h).以上各步水洗后消失的物种也在相应的水洗液中被检测到.

图3 PW11/SBA-15(预水解2 h)样品合成过程中各步骤所取固体产物的IR谱Fig.3 IR spectra of solid products taken from each step in synthesis of PW11/SBA-15 with prehydrolysistime of 2 h(a)(Bu4N)3PW12,(b)(Bu4N)3PW11Si2,(c)solid taken after adding PW11, (d)washing(c)with water,(e)solid taken after crystallization at 40℃, (f)washing(e)with water,(g)solid taken after aging at 80℃, (h)washing(g)with water
由此,我们可将样品的合成过程(见图4)描述如下:首先,在预水解阶段,TEOS水解后形成的低聚态SiO2物种围绕P123胶束组装、聚合,构筑有机-无机聚集体(第I步).PW11加入后,一部分立即与游离态的TEOS作用形成物种(第II步),另一部分则在酸性介质中转化为PW12(第III步).在接下来的40℃晶化过程中,部分物种通过其表面的SiOH与SiO2表面的SiOH缩合,键联到有机-无机聚集体的骨架中,该聚集体在80℃的老化过程中进一步缩合、组装,形成高度有序的介孔结构(第IV步).在高温老化过程中,一些物种(主要是溶液中的游离物种)发生水解成为PW11,并最终转化为PW12(第V步).所有未键联的游离的POM物种均可在水洗或乙醇抽提过程中被除去.

图4 PW11/SBA-15的合成过程Fig.4 Synthesis of PW11/SBA-15
这一反应过程与SiW11/SBA-15的合成[19]相似,但是与SiW11/SBA-15体系相比,在PW11/SBA-15的合成过程中,加入的PW11更易转化为PW12,即使在用量不大、温度不高的情况下也会转化(如图3(c,e)所示).而对SiW11来说,在相似的条件下(比如相同的XW11用量、40℃的晶化过程中),体系中难以检测到SiW12的生成.这可能是由于本文合成中所用的PW11是钠盐,在水中的溶解度很大,并且是配成水溶液后再加入到合成体系中的,所以体系中PW11的浓度较大,相当量的PW11来不及与TEOS反应,就在酸的作用下转化为PW12了.而SiW11/SBA-15合成中所用的SiW11是钾盐,在水中的溶解度较小,溶解较慢,且是以固体的形式直接加入到反应体系中的,这样体系中SiW11的浓度较小.逐渐溶解的SiW11分子优先与TEOS反应,转化为SiW12的量较少.另外,也正是由于体系中PW11的浓度较高,因而对表面活性剂P123的盐析作用较大.我们发现在PW11/SBA-15材料的合成中有更多的粘稠的P123析出,使得样品的洗涤更加困难.
杂化材料的介孔结构可用小角XRD、TEM以及N2吸附进行表征.样品的小角XRD谱示于图5.尽管合成时所用预水解时间的不同对样品中POM的键联负载量没有明显影响,但对杂化材料的介孔结构却影响很大.由图5可见,当预水解时间为0、0.5 h时,没有衍射峰出现(图5的a、b谱),即在此条件下合成的样品为无定形物,没有有序结构.当预水解时间≥1 h时,才有介孔结构形成(图5的c-e谱),且在0.5°-5°范围内,呈现(100)、(110)、(200)三个晶面的衍射峰,表明杂化样品不仅具有SBA-15的六方对称的孔阵列,而且孔阵的长程有序性也比较好.并且随预水解时间的延长,各衍射峰的强度逐渐增加.这些结果都表明足够的预水解时间是形成介孔结构所必须的,也表明预水解时间越长,越有利于有序结构的构造和形成.我们注意到这一点与SiW11/ SBA-15的合成略有不同,在后者的情况下,尽管合成中TEOS也需要预水解,但在一定的预水解时间后,XRD峰强随预水解时间的改变变化不大,即在足够的预水解时间后,介孔结构的构造与预水解时间的长短几乎没有关系.造成这一差别的原因可能是在PW11/SBA-15体系中,PW11的盐析作用更大,对表面活性剂胶束的组装影响更大,所以介孔结构的构造和形成对预水解时间更敏感的缘故.

图5 样品的XRD谱Fig.5 XRD patterns of sampleshybrid samples with prehydrolysis time(h):(a)0,(b)0.5,(c)1, (d)2,and(e)8;(f)pure SBA-15

图6 杂化样品的TEM图像Fig.6 TEM images of hybrid samples (a)sample with prehydrolysis time of 0 h;(b,c)sample with prehydrolysis time of 2 h
杂化样品的TEM图像示于图6.由图可见,预水解时间为0 h的样品没有有序结构(图6a),而预水解时间为2 h的样品则呈现六方有序的孔阵结构(图6b),孔道条纹也清晰有序(图6c),证实了XRD的结果.
样品的N2吸附-脱附等温线和孔径分布图示于图7,孔结构参数则列于表1.可见,预水解时间为0、0.5 h的样品在介孔区没有明显的孔分布(图7B的a、b曲线),其吸附等温线在p/p0为0.65-0.75区间也没有陡峭的毛细凝聚台阶(图7A的a、b)曲线,表面积、孔容较小(表1),再一次表明这些样品结构无序,基本没有介孔.而预水解时间≥1 h的杂化样品,其N2吸附-脱附行为与纯SBA-15相似,具有典型的IV型吸附等温线和H1型滞后环(图7A的c-e曲线),即在中等比压区出现陡峭的毛细凝聚台阶,滞后环的吸、脱附支平行,环形状狭窄,这是中孔材料和孔径均一的圆柱型孔道的特征.孔径分布图(图7B的c-e曲线)也显示样品有大量介孔,孔分布窄,孔径规整均一,这是共缩合法优于二次嫁接法[20]的地方.在二次嫁接法中,POM的引入与介孔结构的构筑不是同时进行,当在已构造完成的介孔孔道中引入较多的POM时,孔道会出现部分堵塞和POM分布不均的现象[20].从表1所显示的孔结构参数则可看出,随预水解时间延长,样品的孔径、孔容减小,壁厚增加,与SiW11/SBA-15的情况相似.这可能是随预水解时间的延长,介孔孔道的构筑趋于完善,更多的POM键联、分布在介孔孔道中,而不在孔壁内部的缘故[19].另外,与纯SBA-15相比,杂化样品的表面积、孔容均明显下降,这主要是由于高分子量的POM的引入导致样品比重增加的缘故,当然也不排除部分有序孔阵的损失所引起的孔参数的下降.
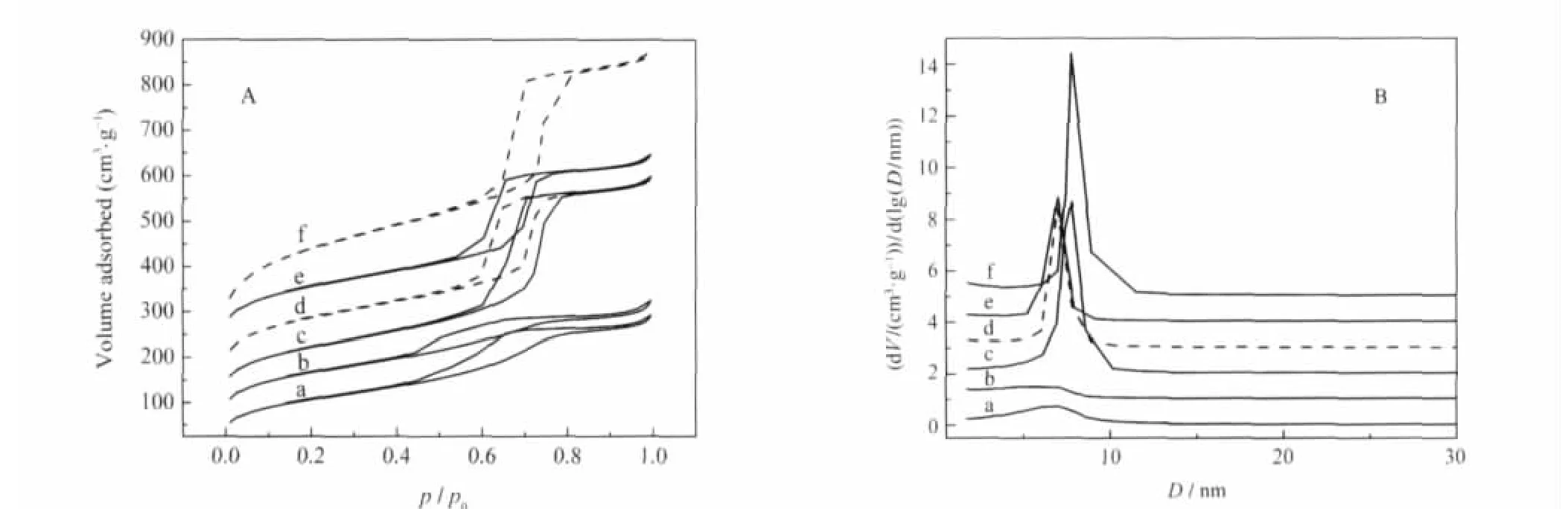
图7 样品的N2吸附-脱附等温线(A)和孔径分布(B)Fig.7 N2adsorption-desorption isotherms(A)and pore size distribution(B)of sampleshybrid samples with prehydrolysis time(h):(a)0,(b)0.5,(c)1,(d)2,(e)8;(f)pure SBA-15;(b),(c),(d),(e),and(f)in Fig.7A were shifted up by 25, 75,125,205,and 200 cm·3g-1,respectively;(b),(c),(d),(e),and(f)in Fig.7B were shifted up by 1,2,3,4,and 5 units,respectively.

表1 样品的结构参数Table 1 Structure parameters of samples
3 结 论
(1)采用共缩合法,在介孔分子筛SBA-15的合成过程中引入一缺位钨磷酸盐PW11,可以合成得到PW11共价键联的介孔杂化材料PW11/SBA-15.其中,不仅多金属氧酸盐的Keggin结构得以保留,并趋于饱和,而且样品仍基本具有六方对称的孔阵列和尺寸均匀的介孔孔道.
(2)TEOS的预水解时间影响着杂化材料的结构.由于盐析作用,在合成体系中PW11的过早加入会干扰模板胶束的组装,进而干扰介孔结构的形成.因此充分的预水解时间是必需的,且随预水解时间的延长,样品的介观有序性增加.
(3)PW11/SBA-15的合成机理与SiW11/SBA-15相似,但由于PW11前驱物的水溶性更大,在体系中的浓度更高,因此合成过程中的转化略有不同,主要是较多的PW11转化为PW12,且PW11对模板剂的盐析作用更大,导致更多的P123析出,不仅导致样品洗涤困难,而且要得到有序度高的样品,需要更长的预水解时间.
1 Vinu,A.;Zakir,K.;Ariga,K.J.Nanosci.Nanotechnol.,2005,5: 347
2 Margolese,D.;Melero,J.A.;Christiansen,S.C.;Chmelka,B.F.; Stucky,G.D.Chem.Mater.,2000,12:2448
3 Corriu,R.J.P.;Mehdi,A.;Reyé,C.;Thieuleux,C.Chem.Mater., 2004,16:159
4 Corriu,R.J.P.;Datas,L.;Guari,Y.;Mehdi,A.;Reyé,C.; Thieuleux,C.Chem.Commun.,2001:763
5 Macquarrie,D.J.;Jackson,D.B.;Tailland,S.;Utting,K.A. J.Mater.Chem.,2001,11:1843
6 Melero,J.A.;Stucky,G.D.;van Grieken,R.;Morales,G.J.Mater. Chem.,2002,12:1664
7 Brunel,D.;Blanc,A.C.;Galarneau,A.;Fajula,F.Catal.Today, 2002,73:139
8 Kim,W.G.;Kim,M.W.;Kim,J.H.;Seo,G.Microporous Mesoporous Mat.,2003,57:113
9 Verhoef,M.J.;Kooyman,P.J.;Peters,J.A.;van Bekkum,H. Microporous Mesoporous Mat.,1999,27:365
10 Nowińska,K.;Fórmaniak,R.;Kaleta,W.;Waclaw,A.Appl.Catal. A-Gen.,2003,256:115
11 Okuhara,T.;Mizuno,N.;Misono,M.Appl.Catal.A-Gen.,2001, 222:63
12 Kaleta,W.;Nowińska,K.Chem.Commun.,2001:535
13 Johnson,B.J.S.;Stein,A.Inorg.Chem.,2001,40:801
14 Schroden,R.C.;Blanford,C.F.;Melde,B.J.;Johnson,B.J.S.; Stein,A.Chem.Mater.,2001,13:1074
15 Guo,Y.;Yang,Y.;Hu,C.;Guo,C.;Wang,E.;Zou,Y.;Feng,S. J.Mater.Chem.,2002,12:3046
16 Chen,L.;Zhu,K.;Bi,L.H.;Suchopar,A.;Reicke,M.;Mathys,G.; Jaensch,H.;Kortz,U.;Richards,R.M.Inorg.Chem.,2007,46: 8457
17 Liu,P.;Wang,H.;Feng,Z.;Ying,P.;Li,C.J.Catal.,2008,256: 345
18 Zhang,R.;Yang,C.J.Nanosci.Nanotechnol.,2007,7(3):1072
19 Zhang,R.;Yang,C.J.Mater.Chem.,2008,18:2691
20 Wang,J.E.;Yang,C.Acta Chim.Sin.,2009,67(4):271 [王金娥,杨 春.化学学报,2009,67(4):271]
21 Kim,G.S.;Hagen,K.S.;Hill,C.L.Inorg.Chem.,1992,31:5316
22 Brevard,C.;Schimpf,R.;Tourne,G.;Tourne,C.M.J.Am.Chem. Soc.,1983,105:7059
23 Agustin,D.;Dallery,J.;Coelho,C.;Proust,A.;Thouvenot,R. J.Organomet.Chem.,2007,692:746
24 Zhao,D.Y.;Feng,J.L.;Huo,Q.S.;Melosh,N.;Fredrickson,G. H.;Chmelka,B.F.;Stucky,G.D.Science,1998,279:548
25 Zhao,D.Y.;Huo,Q.S.;Feng,J.L.;Chmelka,B.F.;Stucky,G.D. J.Am.Chem.Soc.,1998,120:6024
September 4,2009;Revised:October 26,2009;Published on Web:December 22,2009.*
.Email:yangchun@njnu.edu.cn;Tel:+86-25-83598280.
Direct Syntheses and Characterizations of PW11/SBA-15 Mesoporous Hybrid Materials
WANG Jin-E YANG Chun*
(Jiangsu Key Laboratory of Biofunctional Materials,College of Chemistry and Environmental Science, Nanjing Normal University,Nanjing 210097,P.R.China)
The hybrid mesoporous materials,PW11/SBA-15,were synthesized using tetraethoxysilane(TEOS)and lacunary polyoxometalate Na7PW11O39(PW11)by a co-condensation approach in the presence of triblock copolymer EO20PO70EO20(P123)as a template.The resultant materials were characterized by infrared(IR)spectroscopy,UV-Vis diffuse reflectance spectroscopy(UV-Vis/DRS),powder X-ray diffraction(XRD),N2adsorption-desorption,and transmissionelectronmicroscopy(TEM).Results show that the Keggin units are retained perfectly and grafted onto the pore walls of the mesoporous silica by covalent linkage,and an ordered hexagonal packing of channels with homogeneous pore diameters is obtained in the materials.The prehydrolysis time of TEOS influences the construction of the ordered mesostructure significantly.The long-range ordered mesophase increases as the prehydrolysis time increases because of the salt-out effect of PW11,which is associated with solubility of the polyoxometalate precursors.
Mesoporous molecular sieve;Polyoxometalate;Hybrid materials;Na7PW11O39;SBA-15
O643;O611.4;O613.7
The project was supported by the National Natural Science Foundation of China(20473037).
国家自然科学基金(20473037)资助项目
