色谱保留时间在蛋白质组研究中的应用
2010-10-21高友鹤
邵 晨, 高友鹤
(中国医学科学院基础医学研究所,中国协和医科大学基础医学院,生理和病理生理学系,北京100005)
色谱保留时间在蛋白质组研究中的应用
邵 晨*, 高友鹤
(中国医学科学院基础医学研究所,中国协和医科大学基础医学院,生理和病理生理学系,北京100005)
液相色谱与串联质谱联用(LC-MS/MS)技术是蛋白质组学研究中的常见方法。保留时间作为独立于质谱信息的参数已经被用于蛋白质的鉴定和定量工作中。在多肽鉴定领域,多肽的色谱保留时间预测与常规的二级串联质谱数据库搜索算法结合可以提高鉴定的可信度。鉴定的灵敏度也可以通过匹配多次LC-MS实验中具有相同精确质量数和保留时间的峰而提高。另一方面,由于色谱条件的微小改变即会引起保留时间的变化,因此对多次实验结果进行保留时间比对是进行非标记定量的不可或缺的步骤。另外,联合保留时间偏移和质量数信息还可以进行蛋白质翻译后修饰(post-translational modification,PTM)的鉴定。
液相色谱-串联质谱;保留时间预测;保留时间比对;多肽鉴定;非标记定量;翻译后修饰;蛋白质组学
Abstract:Liquid Chromatography coup led with tandem mass spectrometry(LC-MS/MS)has been one of the most popular approaches inproteome analysis.As an independent parameter to mass spectrometry information,peptide retention time has been utilized to facilitate protein identification and quantification.In the field of pep tide identification,the prediction of the retention time combined with routine tandem mass spectrometry database searching methods could help improve the confidence of identification.The sensitivity of identification could also be improved by matching peaks with both the accurate mass and retention time in multiple aligned LC-MS runs.Meanwhile,because small changes of liquid Chromatography conditions lead to variability in retention times unavoidably,retention time alignm ent is crucial to label free quantification.Additionally,post-translational modifications(PTM)could be identified by com bining retention time shifts and mass deviation information.
Key words:liquid Chromatography-tandem mass spectrometry(LC-MS/MS);retention time prediction;retention time alignment;peptideidentification;label-free quantification;posttranslational modification(PTM);proteomics
在蛋白质组的研究中,液相色谱串联一级质谱(liquid Chromatography coupled with mass spectrom etry,LC-MS)或二级质谱(liquid Chromatography coup led with tandem mass spectrometry,LC-MS/MS)是进行蛋白质鉴定和定量分析的常见策略。其主要实验流程为:首先将蛋白质混合物经酶切变为多肽混合物,然后通过一维或二维色谱分离多肽混合物,再用质谱仪鉴定其序列及进行定量分析。用一维色谱分离多肽混合物时,通常是利用反相液相色谱(reversed-phase liquid Chromatography,RPLC)根据多肽的疏水性进行分离,或者根据多肽的电荷特性在RPLC之前再加上强阳离子交换(strong cation exchange,SCX)色谱进行二维分离。在蛋白质组的鉴定和定量分析中,有相当一部分利用了保留时间的信息。本文将对保留时间的预测算法和比对算法及其在蛋白质组研究中的应用进行综述。
我们将首先介绍两项主要技术——保留时间的预测和保留时间的比对——的最新研究进展。随后,将介绍以这两项技术为基础的一系列蛋白质组数据分析算法,包括多肽序列鉴定、翻译后修饰(post-translational m odification,PTM)鉴定和非标记定量。
1 保留时间的预测
以往大多数的蛋白质组鉴定工作都是由质谱的数据出发,比如利用肽段母离子质荷比(mass-tocharge ratio,m/z)和MS/MS谱图中的碎片离子信息等。但是由于多肽混合物的复杂性和噪声信号的影响,鉴定中存在着假阳性和假阴性。多肽的保留时间在近几年被应用到质谱的鉴定中。理想状况下,当色谱分离条件(温度、pH值、流动相组成和固定相)固定时,多肽的保留时间也应保持不变。由于大部分的多肽都不具有实验获得的保留时间信息,所以预测保留时间成为一个很好的替代方法。进行保留时间预测的理论根据是多肽在色谱中的行为与其序列、结构和物理化学性质相关。这里将介绍主要的保留时间预测方法。由于这些预测方法的效果大多是用预测与实际测定的保留时间的相关关系来表示,而相关系数的大小强烈地依赖于进行验证的数据,因此很难通过文献报道比较这些方法的预测效果的好坏。
1.1 根据多肽的序列预测保留时间
最简单的保留时间预测方法是估计每一个氨基酸残基对保留时间的贡献值(系数),再根据多肽的氨基酸组成将它们的保留时间系数加和在一起。这种方法是建立在多肽的保留时间主要是由其氨基酸组成所决定这一假设的基础上的。一种估计氨基酸保留时间贡献的方法[1]是合成特定序列的多肽Ac-G ly-X-X-(Leu)3-(Lys)2-am ide(X为任意20个氨基酸之一),通过测定这些多肽在RPLC的保留时间,从而计算出每一个氨基酸的保留时间(疏水性)系数。在随后的研究中,同一个研究组又引入了一个校正因子,以消除肽段长度对保留时间的影响[2]。除了合成多肽外,很多研究组都利用线性回归模型[3-6],从测定到的已知序列多肽的保留时间中计算每个氨基酸残基的保留时间系数。
近几年,这种通过加和每个氨基酸的保留时间系数计算多肽保留时间的方法又有了新的改进。Petritis等[7]用更加智能化的人工神经网络算法重新计算了氨基酸的保留时间参数。他们计算的新参数与Guo研究组[1,8]的结果有一定的相似性,不同的是,他们认为亮氨酸对保留时间的影响最大,这又与B row ne等[3]的结论相同。Krokhin等[9]发现,除了氨基酸组成和肽链的长度外,多肽N末端的氨基酸残基对保留时间也有很大的影响,因此在他们的预测公式中加入了关于肽段长度和多肽N末端3个残基的校正因子。Kaw akam i等[10]则研究了翻译后修饰对保留时间的影响。他们发现同样是磷酸化修饰,但磷酸化的丝氨酸延迟的保留时间最短,磷酸化的苏氨酸的延迟时间有少许增加,而磷酸化的酪氨酸则会产生较长时间的延迟。这一发现提示保留时间可以应用于鉴定多肽的翻译后修饰及其位点。
1.2 根据多肽的物理化学性质预测保留时间
Petritis等[11]在2006年对他们以前的工作进行了改进,仍然应用人工神经网络算法,但考虑了多肽的物理化学性质信息,其中包括了多肽的长度、序列、氨基酸疏水性、疏水性矩、预测的二级结构以及相邻的氨基酸组合出现的频率。新方法预测的准确率得到了显著的提高,预测与实验的保留时间平均误差为1.5%。由于考虑了多肽序列信息,算法还可以成功地对蛋白质异构体进行区分。
另一种通过物理化学性质预测多肽保留时间的模型被称作定量结构-保留相关关系(quantitative structure retention relationship,QSRR)[12,13]。这个模型表示为:
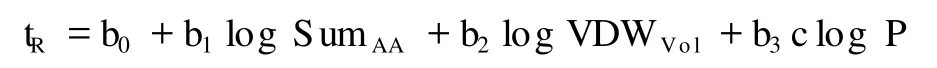
其中:tR为多肽在梯度洗脱下的保留时间;b0为一常数;SumAA是全部氨基酸残基的保留时间系数之和;VDWVol指多肽的范氏体积;clog P则是对数化的正辛醇-水分配系数;b1,b2,b3为上述3个参数的权重,可利用线性回归模型得到。
Asenjo研究组[14-17]一直致力于通过研究多肽表面的氨基酸和色谱柱的疏水性相互作用来预测保留时间。这种方法需要已知多肽结构和建立复杂的数学模型做出相应的预测,而且预测花费的时间较长,并且通量较低。2005年他们研究了一种方法可以只基于氨基酸序列进行预测[14,15]。该方法首先通过多肽的每个氨基酸残基最大可达到的表面积及其暴露在表面的可能性来估计多肽的表面积,再根据其相对分子质量、物理化学性质、二级结构等特征,利用机器学习算法预测多肽的保留时间。
2 保留时间的比对
在蛋白质组特别是临床蛋白质组的研究中,往往需要通过比较很多例的样品来发现潜在的疾病标志物或有特定含义的差异蛋白质,发现方法主要有标记定量和非标记定量。标记定量的方法由于标记试剂种类的限制,只能对数量有限的样品进行定量。而非标记定量方法对每例样品先分别进行LC-MS分析,再将得到的结果先进行保留时间比对再定量分析。由于不需要事先进行样品混合,非标记定量可以进行成百上千例样品的定量,这就克服了标记定量只能比较有限样品数的缺点[18]。把不同次实验产生的LC-MS谱图进行保留时间比对是进行非标记定量的重要步骤。LC-MS谱图比对不但可以消除实验间的色谱分离分析误差,而且使不同时间、不同实验室产生的LC-MS结果进行同时比较成为可能。
进行LC-MS谱图比对的另一个应用是在蛋白质鉴定方面。由于LC-MS/MS实验中,一级质谱扫描到的多肽质谱峰只有少部分会被选择进行二级质谱鉴定得到序列信息,因而在单次的蛋白质组实验中,大量的多肽(或者蛋白质)都不能得到鉴定。如果假设具有相同质量数和保留时间特征的质谱峰所代表的是同一个多肽,通过把同样或相似样品的多次LC-MS谱图比对在一起,只要这个多肽在其中的一次LC-MS/MS中鉴定出来,就可以把其他的实验中具有同样特征的质谱峰也鉴定为这个多肽,这就大大提高了鉴定的多肽和蛋白质的覆盖率和灵敏度[19]。
2.1 保留时间偏差的来源
在色谱分离过程中,相当一部分误差是由色谱柱本身产生的,主要包括色谱柱老化、填充不均匀以及柱内残留的污染物的影响等[20]。即使实验条件控制得很好,这些误差也很难避免。另外,即使是同样的实验条件和样品,更换色谱柱也会造成色谱图的差别。另一方面,一些色谱实验条件(如温度、洗脱梯度等)很难控制也是产生保留时间偏差的主要原因。最后,仪器产生的误差(如死体积和流速的变化、基线漂移等)也会造成很大的影响。因此,在分析不同次的LC-MS数据时,进行保留时间比对是不可或缺的步骤。
2.2 保留时间比对的算法
保留时间比对的算法大致可分为两种:一种是全谱比对算法,即对整个未处理的LC图谱进行全局比对,而几乎不考虑质谱的信息;另一种方法则只比对从总LC-MS谱图中提取出的可能代表多肽的质谱峰,需要将峰的m/z列入计算。由于在比对时只保留了有意义的质谱峰,第二种方法需要计算的数据量较小,但比对的结果非常依赖于多肽峰的检测算法。总的来讲,前一种方法能够处理低m/z分辨率的数据,但计算量较大,不适宜进行过多的LCMS数据的同时比较;而后一种方法往往需要精确的质量数来判断不同实验间代表同一多肽的质谱峰,对质谱仪的要求较高。
2.2.1 LC全谱比对算法
全谱比对算法主要比对不同次LC-MS的总离子流(total ion current,TIC)色谱图,一次实验的TIC谱图可以视为一条在不同的时间点具有不同的总离子流量的曲线。这种曲线在数学上称为连续时间序列。这样,进行两次实验间比对的任务就可以归纳为这样一个数学问题:寻找一个转换函数,使两条曲线之间的距离最小。根据对曲线间距离大小的不同定义,可以用多种动态规划算法,如动态时间规整(dynam ic time w arp ing,D TW)[21]、相关优化偏移(correlation op tim ized w arp ing,COW)[22]、参数时间规整(param etric time w arp ing,PTW)[23]等来求解转换函数。
除了完全基于TIC谱图的方法以外,也有一些算法利用了质谱的信息,即先将总的TIC谱图分成在不同的m/z区间内的子谱图,再进行比对打分。这样可以对复杂度更高的样品进行较好的比对。Listgarten等[24]在使用隐马氏模型进行比对时,发现把每个保留时间点的总离子流量分入4个m/z区间时,既可以提高比对的精确度,也不会带来过大的计算负担。
2.2.2 多肽特征峰比对算法
对TIC谱图进行比对只适合样品的复杂度比较低的情况。当混合物的复杂度较高时,不同的多肽可能在同一时间流出,其色谱峰重叠在一起,在TIC谱图中不能区分。在蛋白质组的研究中,实际关心的只是代表多肽的质谱信号。多肽特征峰比对算法首先检测LC-MS谱图中可能是多肽的质谱峰(可通过具有较高的信噪比,或经由MS/MS鉴定得到序列等特征来判断),称为特征峰(feature),再将可能代表同一多肽的质谱峰匹配起来,比对的目标是使同一多肽的保留时间在历次实验间的误差最小。利用MS/MS的鉴定结果进行特征峰判定最为可靠,但是MS/MS数据不易获得。由于MS/MS扫描速率较慢,在全部可能是多肽的质谱峰中,仅有少部分具有MS/MS鉴定结果。M ueller等[25]报道95%的质谱峰用MS/MS鉴定都可被判定为特征峰,但在他们提取的全部特征峰中仅有10%进行了MS/MS鉴定。
在M ueller小组的方法中,落在相近的m/z和保留时间范围内的特征峰被分为一组,采用局部加权回归散点平滑法估计两次实验间同一组特征峰保留时间的变化。与M ueller等比对全部特征峰的方法相反,Petritis等[7]只选择了6个在多次实验中经常被MS/MS鉴定出序列的多肽作为比对的依据。他们采用遗传算法计算每次实验的线性保留时间转换函数,同时对多次实验的结果进行比对,最后将保留时间归一化到0~1的区间里。
Fischer等[26]采取了折中的办法,首先用岭回归算法根据高可信度的MS/MS数据对两次实验数据进行第一次比对,计算得到一个多项式作为保留时间转换函数。在初始比对的结果上,找到相关度最高的未经MS/MS鉴定的特征峰,然后根据它们的保留时间偏差对多项式进行修正,如此经过数轮迭代,可获得最佳的比对效果。2007年,该小组在原来的算法上做了进一步改进[27],利用多元典型相关分析替代岭回归,解决了原来的算法不具有对称性的问题(对称是指将LC-MS谱图A比对到谱图B上和将谱图B比对到谱图A上获得的结果相同)。
2.2.3 选择恰当的比对算法
最简单的保留时间比对是仅仅通过线性回归来校正不同次实验间的保留时间变化,这种方法虽然比较粗糙,但计算速度最快,健壮性较好。一般情况下,当色谱条件完全相同时,实验间的误差用线性变换来校正即可。然而,大多数的实验都存在着非线性的保留时间误差。计算非线性的保留时间转换函数不仅可以应对洗脱梯度不同的情况,而且比对更为精确。Podw ojski等[28]比较了线性回归方法和两种非线性转换函数,肯定了在对比对的精度要求较高时使用非线性转换函数的必要性。但是非线性算法不仅对计算机的要求较高,转换函数过于复杂时还存在着过拟合的风险,因此需要谨慎地选择。Vandenbogaert等[20]建议首先选择任意一个可以进行非线性比对的软件,用它的比对结果来判断数据是否具有非线性的保留时间误差,再选择恰当的同等级(线性或非线性)的算法进行比对。
不同的比对算法对LC-MS数据本身也有不同的要求。TIC谱图比对需要的计算量最大,当需要比对的实验次数过多或混合物的复杂度较高时,不适宜使用这种算法。特征峰比对算法首先要进行特征提取的步骤,加入这一步骤也带来了额外的误差,尤其是低分辨率质谱仪产生的数据,在进行特征峰提取时将产生较大的误差,对比对的结果有很大的影响。
除了数据本身的特征以外,还应根据比对的目的来选择比对算法。如果是通过比较蛋白质组发现疾病标志物的工作,可以只对保留时间进行较粗略的全局校正以方便定量;而如果是通过保留时间和精确质量数鉴定多肽序列的工作,则对比对的精度要求很高。总而言之,在选择保留时间比对算法时,应根据数据的特征和实际应用的需要,选择最适合而不是最精密复杂的算法,在比对精度、算法健壮性和计算时间之间获得最佳的平衡。
3 利用保留时间进行多肽鉴定
3.1 利用精确质量数和保留时间根据一级质谱数据鉴定多肽
随着质谱技术的发展和质谱仪精度的提高,一些研究试图仅利用酶切多肽的精确质量数和色谱的保留时间鉴定多肽的序列,即只进行LC-MS实验,而不需要再做二级质谱分析。这种方法通常是针对某一特定的组织,通过收集多次LC-MS/MS实验的鉴定结果,建立起这一特定组织的多肽质量和保留时间标签(accurate mass and time tag,AM T tag)数据库。在随后对该组织的实验中,只进行LCMS,而不用二级质谱分析,就可以通过搜索之前建立的数据库来鉴定多肽序列。这种鉴定方法可以大幅度地节约进行二级质谱的时间。由于并不是一级质谱的每一个峰都被选择进行二级质谱分析,而且只有质量好的二级质谱谱图中的多肽会得到正确鉴定,LC-MS/MS的鉴定方法产生了大量的假阴性,且灵敏度不够。只利用LC-MS鉴定的方法可以很好地解决这个问题。应用该方法,低丰度的多肽和高丰度的多肽有同等的机会被鉴定出来。
美国西北太平洋国家实验室在2003年发表文章[29]称将这种方法应用于鉴定耐辐射奇球菌的蛋白质组,使用的仪器是毛细管色谱串联飞行时间质谱(质量精确度<10×10-6(10ppm))。首先,他们根据以前关于耐辐射奇球菌的液相色谱-串联傅里叶变换离子回旋共振质谱和LC-MS/MS实验的结果建立起包含多肽序列、精确质量数和保留时间标签的数据库。在这个数据库中,多肽的保留时间被标准化到一个[0,1]区间里。接下来,对新的LCMS实验谱图中的每一个质谱峰,如果可以在AM T tag数据库中找到唯一的一个序列,使得它们的质量数和保留时间的误差不大于10×10-6和0.05单位时间,那么这个序列就与这个质谱峰匹配。他们报道这种新的鉴定方式具有很高的灵敏度,但是并未考虑该方法的假阳性率。在之后的几年中,他们对这个算法作了一些改进[30,31],如用多肽色谱峰的顶点代替其在质谱中被检测的时间,并应用到多个不同的生物系统[32]和定量蛋白质组的分析[33]中。该研究组于2007年开发出的VIPER软件[34]可以自动地进行LC-MS谱图中特征峰的检测和比对,从AM T tag数据库中找到匹配的记录,从而鉴定多肽序列和进行定量分析。
另一类方法比AM T tag数据库更具有一般性。这类方法不需要事先收集多肽序列和保留时间信息来建立数据库,而是只通过LC-MS比对,直接寻找多次实验中精确质量数和保留时间都十分接近的多肽特征峰,如果这些特征峰中有一部分已经通过MS/MS鉴定出了序列,其他的峰也就随之获得了鉴定。PEPPeR[35]及SuperH irn[25]等都属于这一类型的算法。
3.2 利用保留时间信息进行M S/M S鉴定
在以往的研究中,数据库搜索算法是从MS/MS谱图鉴定多肽的主要方法。该算法的核心思想是将实验谱图和数据库中多肽的理论MS/MS谱图进行比对,并对其匹配程度打分。但这种算法并不完美,其鉴定结果同时存在着假阳性和假阴性的情况。保留时间作为一维新的参数,可以帮助提高MS/MS鉴定的准确性和灵敏度。
一种方法是将保留时间参数和其他评价谱图质量的参数混合成一个新的参数,这个新参数作为唯一的参数,用来决策鉴定是否正确。Strittm atter等[36]提出了一个新的打分函数,是5个参数的加权和,这5个参数分别是预测与实验保留时间误差、质量误差和SEQU EST软件产生的3个评价匹配质量参数。参数的权重通过将一组由已知蛋白质组成的混合物的MS/MS数据作为训练集进行学习而获得。应用新的打分函数,对该混合物的鉴定灵敏度增加了6.5%~9%(分别应用果蝇、大鼠和人类的全蛋白质组数据作为反相数据库检索)。另外,在鉴定人类血浆蛋白质组时,灵敏度增加了16%。
与此相反,另一些研究则把保留时间和谱图匹配的打分参数分开使用。Kaw akam i等[10]把实验和预测的保留时间偏差当作一个预筛选参数,只有保留时间误差在一定的容忍范围内时才能进行随后的多肽匹配步骤。
在Shen等[37]的方法中,当谱图的匹配质量略低于较严格的标准,但高于一个较宽松的标准时,如果预测与实际保留时间的差别很小,同样认定这个MS/MS鉴定是正确的。Pfeifer等[38]也采用了类似的思想,他们用保留时间误差作为过滤条件,从匹配的错误概率大于1%但小于5%的MS/MS谱图中筛选出高可信度的多肽鉴定。在不增加假阳性率的前提下,使鉴定的多肽数目增加了19%。
除了预测的保留时间以外,经验保留时间也被应用于MS/MS鉴定。作者所在课题组[39]在2009年曾通过收集对同一样品重复实验得到的高可信度的MS/MS鉴定和保留时间数据,建立了经验保留时间数据库,从匹配程度较差的MS/MS谱图中筛选高可信度鉴定。虽然经验数据库因只收集了高可信度的经MS/MS鉴定的多肽而使其包含的多肽数量有限,但避免了由于保留时间预测错误造成的误差,与预测的方法形成互补。
4 利用保留时间鉴定蛋白质翻译后修饰
蛋白质的翻译后修饰是蛋白质组研究的重要课题。UN IMOD网站(http://www.un im od.org/)收录的PTM已达数百种之多。传统的检测蛋白质PTM的方法是对样品进行LC-MS/MS分析,然后采用数据库搜索算法来鉴定一种或几种已知的PTM的位点。考虑到同一个氨基酸残基具有被修饰和未被修饰两种质量数不同的状态,而特定的PTM通常在几个特殊的氨基酸残基上出现,鉴定PTM会造成数据库检索空间的数倍乃至数十倍的增加,在检索时间增加的同时,错误匹配机率随之上升。应用数据库检索算法不可能实现同时检索所有蛋白质的PTM的任务。
在蛋白质组样品中,同一个多肽的翻译后修饰和未被修饰的形式往往同时存在。基于这一现象,一些研究组通过对修饰和未修饰两种形式的多肽的母离子m/z、碎片离子和保留时间的相关关系进行PTM的鉴定。
Savitski等[40]发明的M odifiCom b算法利用高质量精度的傅里叶变换质谱数据,可以不受限制地同时鉴定所有存在的PTM,即使是未知的PTM也能够检索到其质量数。该算法主要利用MS/MS提供的多肽碎片离子信息。他们首先将用MASCO T软件从MS/MS谱图中鉴定到的高可信度的未被修饰的多肽序列作为研究的基础,若有另一张MS/MS谱图与基准多肽的MS/MS谱图中有多个(通常定义为4个)碎片离子的m/z相同,或者都相差某个固定的值,则认为这个多肽很可能是基准多肽被修饰之后的形式,它们的质量差可以用来鉴定PTM的类型。他们发现,同一PTM出现在不同的氨基酸残基上会引起不同大小的保留时间偏移,因此应用保留时间可以区分PTM的发生位点。
D asari等[41]根据质量差和保留时间偏移的原理,在低分辨率的质谱仪上鉴定了去酰胺化的多肽(天冬酰胺变为天冬氨酸,或谷氨酰胺变为谷氨酸)。由于质量数相差只有0.984,传统的数据库检索算法不能准确地从低分辨率质谱产生的数据中检索到这一修饰。研究者通过人工合成的多肽,发现修饰和未修饰的多肽在强阳离子交换色谱中的保留时间一致,而在反相色谱分离时去酰胺化的多肽晚3m in流出。利用保留时间差对MS/MS的鉴定结果进行进一步的判定,可以获得93%的PTM鉴定准确率,而通过人工视谱对MS/MS鉴定结果进行判定的准确率只有不足42%。
中国科学院计算技术研究所Fu等[42]于2009年发表论文,提出了一个高效的检索样品中高丰度蛋白质的PTM的方法。和M odifiCom b一样,该算法可以同时检索所有可能存在的PTM。算法只计算多肽的母离子m/z和保留时间偏差,而不考虑MS/MS的信息,因此计算速度更快。首先计算所有谱图两两间的质量差,出现频率很高且质量差在0~100之间则作为可能的候选PTM进入下一步的计算。由于修饰和未修饰的多肽只有一个修饰基团的差别,其物理化学性质比较接近,它们之间的保留时间差理论上是一个固定且较小的数值。基于这个假设,可以利用二元(分别是质量差和保留时间差)混合高斯模型来区分由PTM产生的或随机产生的质量差。在应用于分析糖蛋白质组的数据时,该算法能够比常规的数据检索方法多解释10%的谱图。
现有的研究已经表明,保留时间信息可以帮助研究者更加准确、高效地进行多肽的翻译后修饰的鉴定。但是很少有研究涉及每种PTM给多肽的色谱行为带来的确切影响[10,43],只假设PTM会产生保留时间的较小的恒定的偏移,对保留时间信息的利用还不够充分。如果可以精确地为每种PTM预测可能产生的保留时间偏差,将能够大大提高PTM鉴定的准确度。
5 保留时间比对在定量蛋白质组研究中的应用
准确可靠的定量蛋白质组学研究方法是深入理解不同状态生命的变化、为疾病诊断寻找生物学标记的必要工具。LC-MS比对算法的发展使得多次实验间的非标记定量成为可能,从而加快了生物标志物发现的脚步。
其中一种定量方法是首先从LC-MS谱图中提取多肽的色谱峰,再通过比较这些色谱峰的峰高或峰面积进行定量。一个完整的定量算法一般包括以下几个步骤:(1)MS谱图的预处理;(2)信号的平滑与噪声的去除;(3)特征峰的鉴定并计算其峰高和峰面积;(4)保留时间;(5)误差评估;(6)特征峰分类,寻找生物标记物。Radulovic等[44]开发的软件平台可以进行以上所有步骤的自动化处理。M etA lign软件[45]是另外一个软件,它最多可以对1 000次实验数据进行比对。
提取多肽色谱峰进行定量的方法有一个先天的不足,就是在提取色谱峰的过程中引入了额外的误差,尤其是在处理低分辨率的数据时,误差更为明显。为了避免这一问题,Prakash等[46]直接分析未经过预处理的二维LC-MS图像(分别以时间和m/z为两轴,信号强度显示为点的颜色深浅的信号图)。该算法首先根据名为CHAMS的D TW比对算法对原始的LC-MS图像进行比对。这个比对算法的特点是将峰强度列入考虑范围,特征峰的提取步骤则放在比对之后。他们报道这种方法比先提取特征峰再比对的方法具有更高的特异性和灵敏度。
前面已经对LC-MS比对算法进行了详细的介绍,此处不再赘述非标记定量方法中与保留时间无关的其他技术环节。关于非标记定量方法的介绍可见W ong等[47]和Am erica等[48]的综述。
6 结论
本文介绍了多肽的保留时间信息在蛋白质组数据分析的多个领域中的应用情况。通过AM T tag数据库或比对MS和MS/MS谱图提高蛋白质鉴定灵敏度的方法已经得到了一定程度的应用。利用保留时间也可以鉴定蛋白质的翻译后修饰。另外,在进行非标记定量时,保留时间比对是必不可少的核心步骤。
遗憾的是,利用保留时间进行MS/MS鉴定的研究仍处于算法的发明和验证阶段,实际应用很少。该方法的问题在于,鉴定的准确度强烈地依赖于保留时间预测方法的准确度,而由于预测算法都是通过对有限大小的已知数据集进行学习得到,其可推广性很难得到保证。另外,由于对多肽的色谱行为的了解还不够深入,利用保留时间进行PTM鉴定也处于初级阶段,因此保留时间目前只作为鉴定的辅助信息。尽管存在着一些问题,现有的研究已经证明了保留时间作为独立于质谱数据的参数,可以提高蛋白质鉴定和定量的准确度及效率。随着实验数据的积累和对多肽色谱行为更深入的研究,保留时间将在今后的蛋白质组研究中发挥更重要的作用。
[1] Guo D,M ant C T,Taneja A K,et al.J Chromatogr,1986,359:499
[2] Mant C T,Burke T W L,B lack J A,et al.J Chromatogr,1988,458:193
[3] Browne C A,Bennett H P J,Solom on S.Anal B iochem,1982,124:201
[4] Meek J L.Proc Natl Acad SciUSA,1980,77:1632
[5] Meek J L,Rossetti Z L.J Chromatogr,1981,211:15
[6] Sakamoto Y,Kaw akam i N,Sasagawa T.J Chromatogr,1988,442:69
[7] Petritis K,Kangas L J,Ferguson P L,et al.Anal Chem,2003,75(5):1039
[8] Guo D,M ant C T,Taneja A K,et al.J Chromatogr,1986,359:518
[9] Krokhin O V,Craig R,Spicer V,et al.mol Cell Proteomics,2004,3(9):908
[10] Kawakami T,Tateishi K,Yam ano Y,et al.Proteomics,2005,5(4):856
[11] Petritis K,Kangas L J,Yan B,et al.Anal Chem,2006,78(14):5026
[12] Baczek T,W iczling P,M arszallM,et al.J Proteome Res,2005,4(2):555
[13] Kaliszan R,Baczek T,Cim ochow ska A,et al.Proteomics,2005,5(2):409
[14] Salgado J C,Rapaport I,Asenjo J A.J Chromatogr A,2005,1098:44
[15] Salgado J C,Rapaport I,Asenjo J A.J Chromatogr A,2005,1075:133
[16] Salgado J C,Rapaport I,Asenjo J A.J Chromatogr A,2006,1107:120
[17] Salgado J C,Rapaport I,Asenjo J A.J Chromatogr A,2006,1107:110
[18] Old W M,Meyer-Arendt K,Aveline-Wolf L,et al.mol Cell Proteomics,2005,4(10):1487
[19] Li X J,Yi E C,Kemp C J,et al.Mol Cell Proteomics,2005,4(9):1328
[20] VandenbogaertM,Li-Thiao-Te S,Kaltenbach H M,et al.Proteomics,2008,8(4):650
[21] Bylund D,Danielsson R,Malmquist G,et al.J Chromatogr A,2002,961(2):237
[22] Christin C,Sm ilde A K,Hoefsloot H C,et al.Anal Chem,2008,80(18):7012
[23] Eilers P H.Anal Chem,2004,76(2):404
[24] Listgarten J,Neal R M,Roweis S T,et al.Bioinformatics,2007,23(2):e198
[25] M ueller L N,Rinner O,Schmidt A,et al.Proteomics,2007,7(19):3470
[26] Fischer B,Grossm ann J,Roth V,et al.B ioinformatics,2006,22(14):e132
[27] Fischer B,Roth V,Buhmann J M.BMC B ioinformatics,2007,8(Supp l10):S4
[28] Podwojski K,Fritsch A,Cham rad D C,et al.Bioinformatics,2009,25(6):758
[29] Strittmatter E F,Ferguson PL,Tang K,et al.J Am Soc mass Spectrom,2003,14(9):980
[30] Zimm er J S,M onroe M E,Q ian W J,et al.mass Spectrom Rev,2006,25(3):450
[31] Kiebel G R,Auberry K J,Jaitly N,et al.Proteom ics,2006,6(6):1783
[32] Adkins J N,Monroe M E,Auberry K J,et al.Proteom ics,2005,5(13):3454
[33] M anes N P,Estep R D,Mottaz H M,et al.J Proteom e Res,2008,7(3):960
[34] M onroe ME,Tolic N,Jaitly N,et al.B ioinform atics,2007,23(15):2021
[35] Jaffe J D,M ani D R,Lep tos K C,et al.Mol Cell Proteom ics,2006,5(10):1927
[36] Strittm atter E F,Kangas L J,Petritis K,et al.J Proteom e Res,2004,3(4):760
[37] Shen Y,Kim J,Strittm atter E F,et al.Proteom ics,2005,5(15):4034
[38] Pfeifer N,Leinenbach A,Huber C G,et al.J Proteom e Res,2009,8(8):4109
[39] Sun W,Zhang L,Yang R,et al.Rap id Comm un Mass Spectrom,2009,23(1):109
[40] Savitski M M,N ielsen M L,Zubarev R A.mol Cell Proteom ics,2006,5(5):935
[41] Dasari S,W ilm arth P A,Rustvold D L,et al.J Proteom e Res,2007,6(9):3819
[42] Fu Y,J ia W,Lu Z,et al.BMC B ioinform atics,2009,10(Supp l1):S50
[43] Kim J,Petritis K,Shen Y,et al.J Chromatogr A,2007,1172(1):9
[44] Radulovic D,Jelveh S,Ryu S,et al.mol Cell Proteom ics,2004,3(10):984
[45] Lomm en A.Anal Chem,2009,81(8):3079
[46] Prakash A,M allick P,Whiteaker J,et al.mol Cell Proteom ics,2006,5(3):423
[47] W ong J W,Sullivan M J,Cagney G.B rief B ioinform,2008,9(2):156
[48] Am erica A H,Cordew ener J H.Proteom ics,2008,8(4):731
Application of peptide retention time in proteome research
SHAO Chen*,GAO Youhe
(Department of Physiology and Pathophysiology,Institute of Basic Medical Sciences,Chinese Academy of Medical Sciences,School of Basic Medicine,Peking Union Medical College,Beijing 100005,China)
O658
A
1000-8713(2010)02-0128-07
*通讯联系人:邵 晨,助理研究员,主要研究方向为蛋白质组学和生物信息学.Tel:(010)65296407,E-m ail:scshaochen@126.com.
中国医学科学院基础医学研究所院(所)长基金项目(No.2009PY05)、国家自然科学基金杰出青年基金项目(No.30725009)、国家自然科学基金项目(No.30870502)、北京市自然科学基金项目(No.5072037)和高等学校博士学科点专项科研基金项目(No.20070023021).
2009-12-24
DO I:10.3724/SP.J.1123.2010.00128
