HPLC测定市售青黛中靛蓝和靛玉红的含量
2010-10-16白宗利任玉珍陈彦琳杜杰梁焕彭秀娟周林田壮屈效源
白宗利,任玉珍,陈彦琳,杜杰,梁焕,彭秀娟,周林,田壮,屈效源
(中国药材公司,北京 100195)
HPLC测定市售青黛中靛蓝和靛玉红的含量
白宗利*,任玉珍,陈彦琳,杜杰,梁焕,彭秀娟,周林,田壮,屈效源
(中国药材公司,北京 100195)
目的:建立青黛中靛蓝和靛玉红含量的测定方法。方法:采用HPLC定量分析,色谱柱为Diamonsil C18(150mm×4.6mm,5μm),靛蓝流动相为甲醇-水(60∶40),流速为1.0mL·min-1,检测波长为610nm;靛玉红流动相为甲醇-水(70∶30),流速为1.0mL·min-1,检测波长为 292nm。结果:靛蓝在 0.029 6~0.296μg(r=0.999 9)与峰面积呈良好线性关系,靛玉红在0.010 6~0.127 2μg(r=0.999 9)与峰面积呈良好线性关系。结论:HPLC操作简便、结果可靠、重现性好、专属性强,可用于青黛中有效成分含量的测定。
青黛;靛蓝;靛玉红;高效液相色谱法
青黛为爵床科植物马蓝Baphicacanthus cusia(Nees)Bremek.、蓼科植物蓼蓝Polygonum tinctoriumAit.或十字花科植物菘蓝Isatis indigoticaFort.的叶或茎叶经加工制得的干燥粉末、团块或颗粒。具有清热解毒,凉血消斑,泻火定惊的功能。用于温毒发斑,血热吐衄,胸痛咳血,口疮,痄腮,喉痹,小儿惊痫[1]。青黛的主要有效成分是靛蓝和靛玉红,《中国药典》2005年版一部中分别采用紫外-可见分光光度法和薄层色谱扫描法对其含量进行测定[2],但这些方法存在着操作繁琐、重现性差等问题。我们通过修订 《北京市中药炮制规范》,参考 《中国药典》2005年版一部蓼大青叶项下 “含量测定”[3]和 《中国药典》2010年版一部青黛项下 “含量测定”中靛玉红的含量测定[1],采用高效液相色谱法分别测定了9批市售青黛样品中靛蓝和靛玉红的含量,现将结果报道如下。
1 仪器与试药
1.1 仪器
waters2695液相色谱仪,DAD2996检测器;岛津uv2550紫外分光光度计;KQ3200超声波清洗器(昆山市超声仪器有限公司);DENVER电子分析天平(1/10万,北京赛多利斯仪器系列有限公司)。
1.2 试药
靛蓝对照品(批号110716-200509,供含量测定用);靛玉红对照品(批号110717-200204,供含量测定用),均购自中国药品生物制品检定所。
1.3 试剂与样品
三氯甲烷、水合氯醛、甲苯、丙酮为分析纯;甲醇为色谱纯;水为娃哈哈纯净水;硅胶G薄层板(青岛海洋化工厂)。
青黛样品分别从北京市5家中药饮片厂和北京市知名药店收集,共9批,编号及来源详见表1。经北京市卫生学校金世元教授鉴定为爵床科植物马蓝Baphicacanthus cusia(Nees)Bremek.、蓼科植物蓼蓝Polygonum tinctoriumAit.或十字花科植物菘蓝Isatis indigoticaFort.的叶或茎叶经加工制得的干燥粉末。

表1 青黛样品编号及来源
2 方法与结果
2.1 青黛中靛蓝含量测定
2.1.1 色谱条件 色谱柱:Diamonsil C18(150mm×4.6mm,5μm);流动相:甲醇-水(60∶40);柱温:30℃;流速:1mL·min-1;检测波长:610nm;进样量:10μL;理论板数按靛蓝峰计算应不低于1 800。2.1.2对照品溶液的制备 取靛蓝对照品3.7mg,精密称定,置250mL量瓶中,加2%水合氯醛的三氯甲烷溶液(取水合氯醛,置硅胶干燥器中放置24h,称取2.0g,加三氯甲烷至100mL,放置,出现浑浊,以无水硫酸钠脱水,滤过,即得。)约200mL,超声处理(功率250W,频率33kHz)1.5h,取出,放冷至室温,加2%水合氯醛的三氯甲烷溶液至刻度,摇匀,即得浓度为0.014 8mg·mL-1的对照品溶液。靛蓝对照品HPLC图见图1。

图1 靛蓝对照品HPLC图
2.1.3 供试品溶液的制备 取青黛细粉约25mg,精密称定,置25mL量瓶中,加2%水合氯醛的三氯甲烷溶液约 20mL,超声处理(功率 250W,频率33kHz)1.5h,取出,放冷,加2%水合氯醛的三氯甲烷溶液至刻度,摇匀,滤过,取续滤液,即得。青黛样品HPLC图见图2。

图2 青黛样品HPLC图
2.1.4 线性关系考察 精密吸取对照品溶液2,5,10,12,15,20μL,注入液相色谱仪,按上述色谱条件测定峰面积;以靛蓝进样量为横坐标,以色谱峰面积为纵坐标作线性回归,得回归方程:Y=350 218 6.2X-322 56.0(r=0.999 9),结果表明,靛蓝在0.029 6~0.296μg与峰面积呈良好线性关系。
2.1.5 精密度试验 精密吸取上述配制的靛蓝对照品溶液10μL,重复进样6次,测定峰面积,结果RSD=0.5%;精密吸取上述配制的供试品溶液10μL,重复进样 6次,测定靛蓝峰面积,结果RSD=0.7%,表明本方法精密度良好。
2.1.6 稳定性试验 精密吸取同一供试品溶液10μL,分别于0,2,4,6,8,12h时进样,测定靛蓝峰面积,结果RSD=1.6%,表明样品溶液在12h内基本稳定。
2.1.7 重复性试验 取同一批青黛样品6份,按供试品溶液的制备方法制备,照上述色谱条件各进样10μL测定,结果靛蓝的平均含量为1.86%,RSD=0.5%,说明本方法重复性良好。
2.1.8 回收率试验 取已知靛蓝含量的青黛(含量为1.86%)12.5mg,精密称定,共6份,置25mL量瓶中,分别加入浓度为11.98μg·mL-1的靛蓝对照品溶液,按供试品溶液的制备方法制备,照上述色谱条件测定,计算回收率。结果见表2。

表2 靛蓝加样回收率试验
2.1.9 检测限和定量限试验 采用信噪比法,先进1针空白溶剂,测出噪音,进已知浓度靛蓝对照品,根据其对应的峰高值,将对照品溶液稀释至约3倍信噪比浓度,测得靛蓝的检测限为0.710 4μg·mL-1;同样将对照品稀释至约10倍信噪比的浓度,重复进样5~6次,测得靛蓝的定量限为2.368μg·mL-1。
2.1.10 样品靛蓝含量测定结果 取9批青黛样品,按上述方法进行靛蓝含量测定,结果(以干燥品计)见表4。
2.2 青黛中靛玉红含量测定
2.2.1 色谱条件 色谱柱:Diamonsil C18(150mm×4.6mm,5μm);流动相:甲醇-水(70∶30);柱温:25℃;流速:1mL·min-1;检测波长:292nm;进样量:10μL;理论板数按靛玉红峰计算应不低于3 000。
2.2.2 对照品溶液的制备 取靛玉红对照品2.65mg,精密称定,置50mL量瓶中,加N,N-二甲基甲酰胺约45mL,超声处理(功率250W,频率33kHz)至溶解完全,取出,放冷至室温,加N,N-二甲基甲酰胺至刻度,摇匀;精密量取10mL,置100mL量瓶中,加N,N-二甲基甲酰胺至刻度,摇匀,即得0.005 3mg·mL-1的对照品溶液。靛玉红对照品HPLC图见图3。
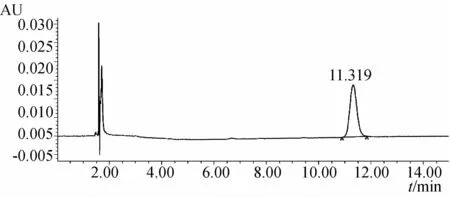
图3 靛玉红对照品HPLC图
2.2.3 供试品溶液的制备 取本品适量,研细,取约50mg,精密称定,置25mL量瓶中,加N,N-二甲基甲酰胺约20mL,超声处理(功率250W,频率33kHz)30min,取出,放冷,加 N,N-二甲基甲酰胺至刻度,摇匀,滤过,取续滤液,即得。青黛样品HPLC图见图4。
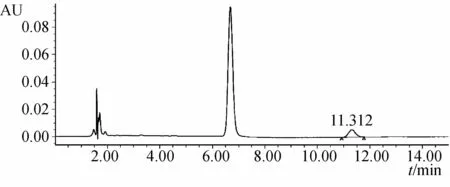
图4 青黛样品HPLC图
2.2.4 线性关系考察 精密吸取对照品溶液2,4,8,16,20,24μL,注入液相色谱仪,按上述色谱条件测定峰面积;以靛玉红进样量为横坐标,以色谱峰面积为纵坐标作线性回归,得回归方程Y=276 21X-175 72.0(r=0.999 9),结果表明,靛玉红在0.010 6~0.127 2μg与峰面积呈良好线性关系。
2.2.5 精密度试验 精密吸取上述配制的靛玉红对照品溶液10μL,重复进样6次,测定峰面积,结果RSD=0.3%;精密吸取上述配制的供试品溶液10μL,重复进样6次,测定靛玉红峰面积,结果RSD=1.0%,表明本方法精密度良好。
2.2.6 稳定性试验 精密吸取同一供试品溶液10μL,分别于0,2,4,6,8,12h进样,测定靛玉红峰面积,结果RSD=0.7%,表明样品溶液在12h内基本稳定。
2.2.7 重复性试验 取同一批青黛样品6份,按供试品溶液的制备方法制备,照上述色谱条件各进样10μL测定,结果靛玉红的平均含量为0.108 3%,RSD=1.4%,说明本方法重复性良好。
2.2.8 回收率试验 取已知靛玉红含量的青黛(含量为0.11%)25mg,精密称定,共6份,置25mL量瓶中,分别加入浓度为0.005 3mg·mL-1的靛玉红对照品溶液,按供试品溶液的制备方法制备,照上述色谱条件测定,计算回收率,结果见表3。

表3 靛玉红加样回收率试验
2.2.9 检测限和定量限试验 采用信噪比法,先进1针空白溶剂,测出噪音,进已知浓度靛玉红对照品,根据其对应的峰高值,将对照品溶液稀释至约3倍信噪比浓度,测得靛玉红的检测限为 0.361μg·mL-1;同样将靛玉红对照品稀释至约10倍信噪比的浓度,重复进样5~6次,测得靛玉红的定量限为1.06μg·mL-1。
2.2.10 样品靛玉红含量测定结果 取9批青黛样品,按上述方法进行靛玉红含量测定,结果(以干燥品计)见表4。

表4 9批次青黛样品中靛蓝和靛玉红含量测定/%
3 讨论
《中国药典》2005年版一部青黛项下规定靛蓝含量不少于2.0%,靛玉红含量不少于0.13%。《中国药典》2010年版虽然修订了靛蓝的含量测定方法,但有关限度未变。本文测定结果表明,收集的9批市售青黛样品中,有1批样品靛蓝含量小于2.0%,有6批样品靛玉红含量小于0.13%,说明目前市场流通使用的青黛质量问题严重,应引起有关部门的重视。 实验表明,HPLC操作简便、结果可靠、重现性好、专属性强,可用于青黛中有效成分含量的测定。
[1]国家药典委员会.中国药典(一部)[S].北京:中国医药科技出版社,2010:185.
[2]国家药典委员会.中国药典(一部)[S].北京:化学工业出版社,2005:138-139.
[3]国家药典委员会.中国药典(一部)[S].北京:化学工业出版社,2005:253.
Determ ination of Indigo and Indirubin in Indigo Naturalis by HPLC
Bai Zongli,Ren Yuzhen,Chen Yanlin,Du jie,Liang Huan,Peng Xiujuan,Zhou Lin,Tian Zhuang,Qu Xiaoyuan
(China National Corp.of Traditional&Herbal Medicine,Beijing100195,China)
Objective:To determine the contents of indigo and indirubin in indigo naturalis.Methods:The contents of indigo and indirubin were determined by HPLC.The chromatographic column was Diamonsil Cl8(150mm×4.6mm,5μm).The mobile phase was methanol-water(60∶40)and the flow rate was 1.0mL·min-1with detection wavelength of 610nm for indigo.The mobile phase was methanol-water(70∶30)and the flow rate was 1.0mL·min-1with detection wavelength of 292nm for indirubin.Results:The linear correlation of the indigo was good in the range of 0.029 6~0.296μg(r=0.999 9).The Linear correlation of the indirubin was good in the range of 0.010 6~0.127 2μg(r=0.999 9).Conclusion:HPLC method is simple and the result is reliable with good repeatability and can be used for the determination of the components in indigo naturalis.
Indigo naturalis;Indigo;Indirubin;HPLC
*白宗利,E-mail:baizl@sino-tcm.com
2010-03-18)
