水分子配位对叶绿素a氧化还原势与红外光谱的影响
2010-10-14王国营欧家鸣王瑞丽
胡 琼 王国营 欧家鸣 王瑞丽
(云南师范大学物理与电子信息学院,昆明 650092)
水分子配位对叶绿素a氧化还原势与红外光谱的影响
胡 琼 王国营 欧家鸣 王瑞丽*
(云南师范大学物理与电子信息学院,昆明 650092)
以光系统I反应中心电子传递链上的辅助叶绿素a为目标,采用密度泛函理论中的B3LYP方法,结合三种基组,系统计算了该叶绿素及其两种配位分子模型在气相、模拟蛋白质环境和水中的氧化还原势和离解能;同时计算了这三种模型在气相中的几何结构、红外光谱及其13C、15N和2H的同位素标记谱.计算及分析结果表明:水分子配位引起镁离子偏离叶绿素a的卟啉环平面中央,导致以镁原子为中心的键角减小,Mg—N键长增长;而天冬酰胺对配位的水分子施加氢键影响后,使得Mg—N键进一步增长,镁离子与水分子中氧原子的配位键Mg—O键长减小,离解能增加,合成分子的氧化还原势减少;另外,分子的氧化还原势和配位键离解能随着相对介电常数的增加以及计算基组的增大而减小;三种分子模型的羰基(C=O)和卟啉环上C=C键的特征振动频率差值小于7 cm-1,而同位素标记引起其峰位变化量的差值小于3 cm-1.该计算为研究光合反应中心电子传递链上叶绿素a的作用与功能提供理论参考依据.
密度泛函理论; 叶绿素a; 氧化还原势; 离解能; 红外光谱; 基组
Abstract:In the reaction center of photosystem I the accessory electron transfer cofactors are two monomeric chlorophyll-a molecules that are ligated to two water molecules.To study the effect of water ligation on the redox potential and vibrational properties of chlorophyll-a,we built three molecular models of water ligation of chlorophyll-a based on the X-ray crystal structure of photosystem I.Then,we systematically calculated the geometries,vibrational frequencies,bond dissociation energies,and redox potentials of these models using density functional theory.The calculations were conducted in the gas phase,water,and a simulated protein environment.In addition,three different basis sets were employed to investigate the influence of the basis set on the calculation results.15N,2H,and13C labeled spectra of the models in the gas phase were also calculated.Our results show that the water ligand causes the Mg ion of chlorophyll-a to move away from the center of the porphyrin ring so that the Mg—N bond lengths increase and the Mg centered angles decrease.When a nearby amino acid,asparagine(ASNB591),provides a hydrogen bond to the water that is axial ligand to the chlorophyll-a,these changes increase further.Additionally,the Mg—O bond distance decreases,the dissociation energy increases,and the redox potential also decreases.Furthermore,the redox potentials of the molecules and their bond dissociation energies decrease as the relative dielectric constant of the media and the basis sets increase.However,differences in the frequencies of the corresponding carbonyl groups and the C=C vibrations of the porphyrin ring in the three models are less than 7 cm-1,and the differences in frequency shift upon isotope labeling between the models are less than 3 cm-1.These results provide useful information for further studies of the structural and functional properties of chlorophyll-a in the photosynthetic reaction center.
Key Words: Density functional theory; Chlorophyll-a; Redox potential; Dissociation energy;Infrared spectrum; Basis set
叶绿素a是一种重要的光合色素,尤其在I类光合反应中心,它既作为电子传递链上的供体,又是电子传递的受体,在光能转换到电能的过程中具有举足轻重的地位[1].
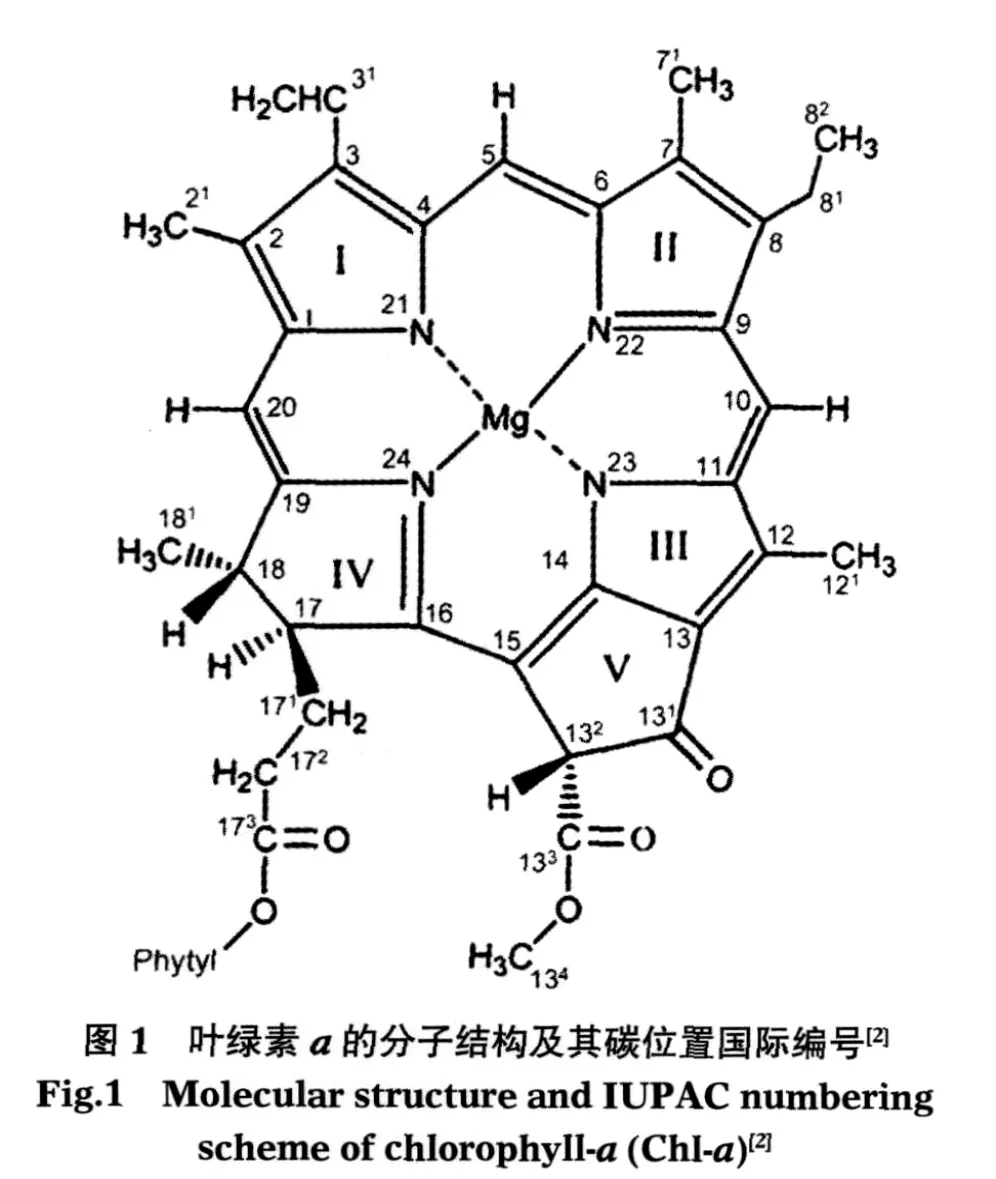
叶绿素a的分子结构及其碳位置国际编号如图1所示[2].完整叶绿素a含有137个原子,其中央含有一个镁离子,在复合结构中,该镁离子通常与其它分子结合而形成轴向配位键,从而对叶绿素a的结构和特性产生影响.叶绿素a含有三个羰基,分别是C131=O、C133=O和C173=O.这三个羰基具有较大的偶极动量,给出强度较高的振动特征峰位,在叶绿素a的红外光谱研究中非常重要[3-4].
目前,光合系统的晶体结构研究表明,光系统I反应中心含有3对叶绿素a,排列在非常对称的两条路径上[5-6].然而这6个叶绿素a各自所处的蛋白质环境均不相同.初级电子供体P700由叶绿素a和叶绿素a的同质异构体叶绿素a′组成,分别配位于组氨酸,初级电子受体A0是两个配位于蛋氨酸的叶绿素a.本文涉足的是第三对叶绿素a,又名辅助叶绿素A,位于P700和A0之间,分别配位于两个水分子.另外,这三对叶绿素a的配位体都受到来自其它蛋白质的不同强度的氢键束缚,这些配位体及其蛋白质环境对叶绿素a在光合反应中心结构与功能的影响一直是人们探寻基于光合作用原理的人造电子器件的研究热点[1,6].
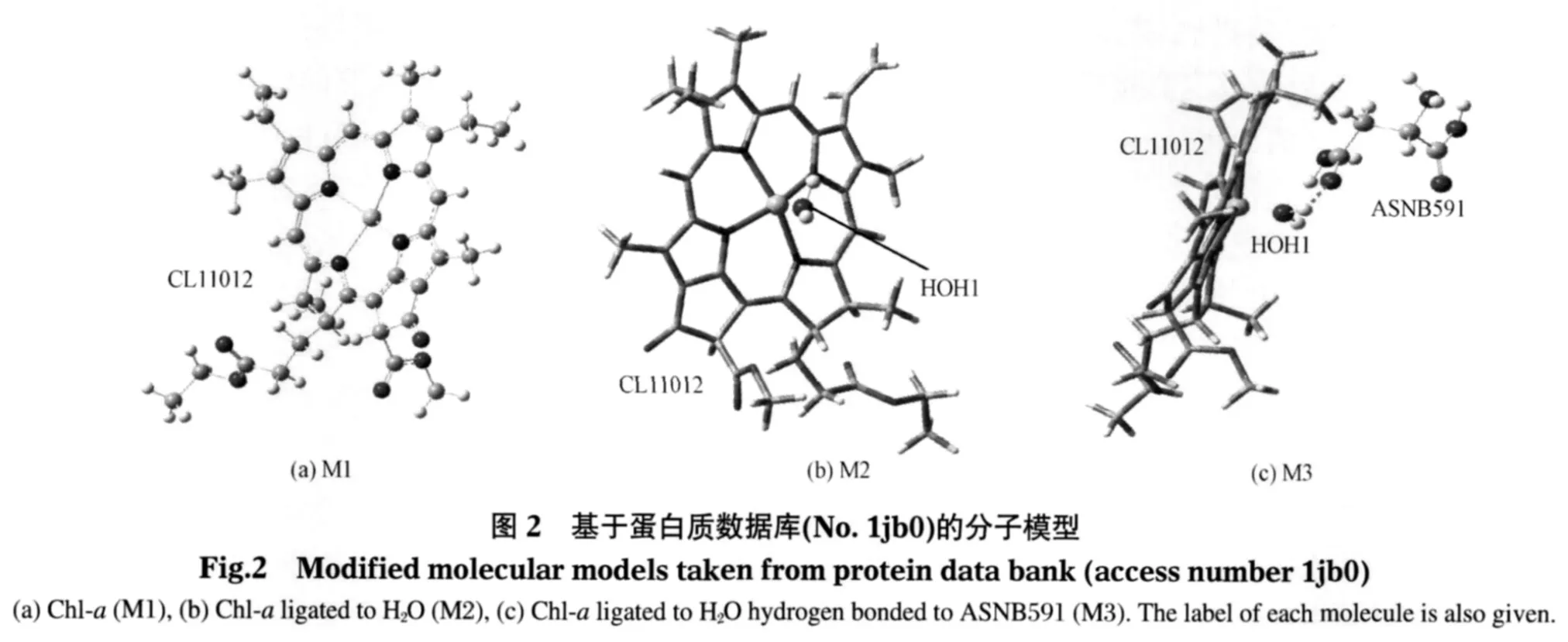
氧化还原势是研究电子传递体特性的重要物理参数,而红外光谱是研究分子结构的重要方法之一.人们发现,采用基因点定位法改变构成P700的两个叶绿素a的配位体,将引起P700的空间构象和氧化还原势的改变[7-8],而改变A0的配位体蛋氨酸引起其可见光谱的改变和传递电子速度的减慢[9].然而,对配位于水的辅助叶绿素A的基因点定位实验研究还未见报道,这两个叶绿素是否参与电子传递尚存争议[10-11].近年来,随着计算科学的高速发展,人们已用含时密度泛函理论研究了由介质所形成的配位键和氢键对叶绿素a在基态和不同电子激发态光谱特性的影响[12-15],采用密度泛函理论研究叶绿素a的红外光谱特性、不同配位体对叶绿素a的性能影响以及其可见光谱特性已有报道[2,16-19],但采用的分子模型和计算方法均有限,而对光合反应中心水分子配位对辅助叶绿素A红外光谱特性影响的理论计算研究还未见报道.为此,我们以光系统I的蛋白质库中电子传递链上的分子为基础建立了三种分子模型:叶绿素a(简写M1),叶绿素a配位于水分子(简写M2),提供叶绿素a配位键的水分子受到来自天冬酰胺的氢键束缚(简写M3),如图2所示.
我们采用密度泛函理论B3LYP方法,结合6-31G(d)、6-311+G(d)和 6-311+G(2d,2p)基组,系统计算了这三种分子模型在气相、模拟蛋白质环境和水中配位键的离解能、氧化还原势及其在气相中的几何结构、红外光谱和15N、13C和2H的同位素标记谱.目的在于探索水分子配位和蛋白质环境对叶绿素a的结构、氧化还原势、光谱特性及其功能以及计算基组和介质的影响,为研究光合反应中心电子传递链上叶绿素a的作用与功能提供理论参考依据.
1 计算方法
三种分子模型的原初结构均取自光系统I蛋白质数据库(编号1jb0),该蛋白质库来源于0.25 nm X衍射晶体结构[1].为减少计算时间,模型中的叶绿素a剪切在Phytyl位置(图1)并以甲基取代叶绿基,因而含有85个原子.为保持分子中性,我们利用GaussView 4.1[19]将模型M3中两性离子态的天冬酰胺的氨基上的一个H原子剪接到羧基的氧原子上(图2(c)),剪接过程中保持该分子处于最低能量状态.图2(a,b,c)分别给出取自蛋白质数据库并经剪切后的三种分子模型结构以及各个亚分子在蛋白质库中的编号.所有理论计算均使用Gaussian 03软件D.01版本[20].几何优化和频率计算均采用密度泛函理论中的B3LYP方法结合6-31G(d)基组进行,介质采用气相.根据理论计算要求,频率计算是在对分子进行几何优化的基础上,采用相同方法计算.能量计算同样选用频率计算中所用的分子模型和方法,结合6-31G(d)、6-311+G(d)和 6-311+G(2d,2p)三种基组完成.为了进一步理解介质对分子物理化学特性的影响,我们采用集成方程式和极化连续介质模型(IEFPCM)[21]分别计算了三种分子模型在介质气相(相对介电常数ε=1.0)、模拟蛋白质环境和水中的氧化还原势和离解能.模拟蛋白质环境反映的是介质的极化特性和介质中蛋白质的空间取向,其相对介电常数一般在2至16之间[16,22],在此选用ε=4.0.
2 结果及讨论
2.1 几何结构
表1给出采用B3LYP/6-31G(d)方法所得出的三种分子模型的基态和阳离子态在气相(ε=1.0)中的主要几何优化参数.由表1明显看出:水分子配位引起镁离子偏离叶绿素a的四吡咯环构成的平面,导致以镁原子为中心的键角减小,基态/阳离子态的最大减小量为1.382°/1.636°,基态的Mg—N键长增长在1.8-3.7 nm之间,阳离子态的Mg—N键长增长在2.9-3.7 nm之间.而天冬酰胺对水分子施加氢键影响后,镁离子的偏离程度进一步增加;与M1相比,基态和阳离子态以Mg原子为中心的键角分别减小在 1.359°-2.117°和 2.160°-2.571°之间,基态的Mg—N键长增长在3.1-5.7 nm之间,阳离子态的Mg—N键长增长在4.0-5.3 nm之间;同时使镁离子与水分子中的氧原子的距离,即Mg—O配位键的键长减小.而且,所有模型中,无论是基态还是阳离子态,最短的Mg—N键是Mg—N23键,最长的是Mg—N24键.以M1模型为例,其基态/阳离子态中Mg—N23和Mg—N24的键长分别是0.2019/0.2014 nm和0.2151/0.2138 nm.另外,由表1还看出,由于所有模型中的羰基C=O键离中央镁离子相对较远,受到配位体的影响甚小,键长的变化仅在0.0001 nm以内.几何参数的改变直接关系到分子氧化还原势及其红外光谱的变化.
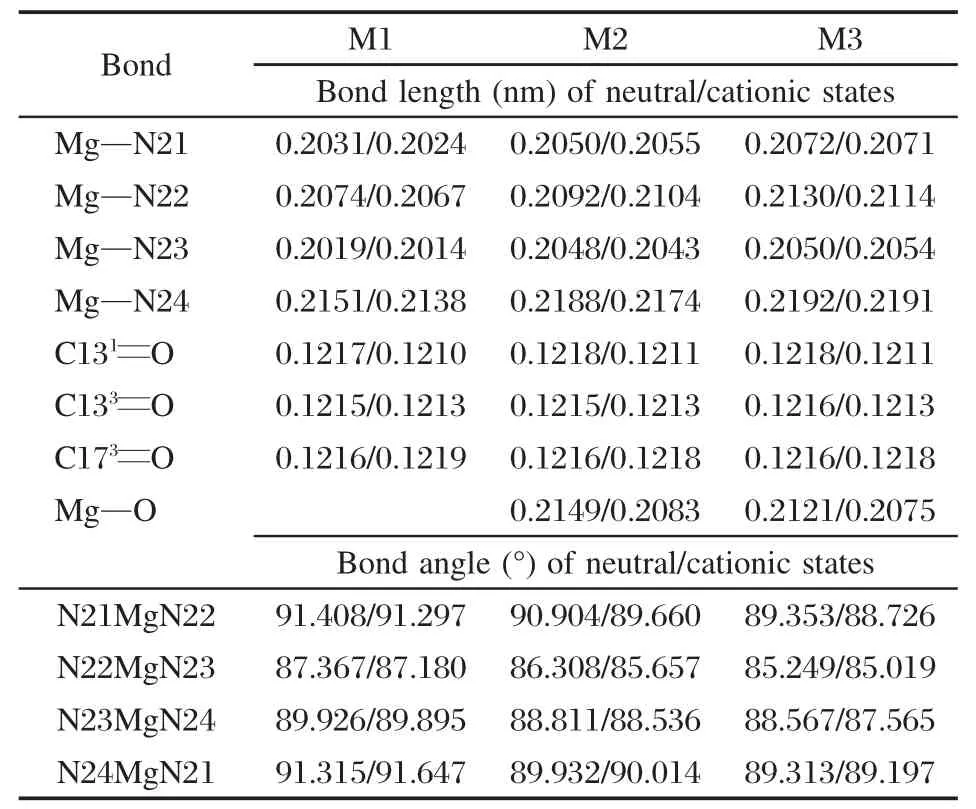
表1 计算出的三种分子模型的基态/阳离子态在气相中的主要几何优化参数Table 1 Calculated geometry optimized parameters for three molecular models at neutral/cationic states in the gas phase
2.2 离解能
叶绿素与轴向配位体形成的配位键的束缚强度可由其离解能△E来量度,通过下式估算[15]:

表 2给出采用 B3LYP方法结合 6-31G(d)、6-311+G(d)和6-311+G(2d,2p)三种基组计算出的基态分子模型M2和M3在介质气相(ε=1.0)、模拟蛋白质环境(ε=4.0)和水(ε=78.4)中的离解能.模型 M3 配位键的离解能是由叶绿素a和水分子与天冬酰胺复合体的能量之和减去模型M3的能量而得.
表2的数据表明,在不同基组和不同介质下,所有计算出的模型M3的离解能均高于相应的模型M2的离解能.由于在模型M3中,叶绿素a的配位体水分子受到来自天冬酰胺的氢键束缚的影响,Mg—O配位键的键长减小(见表1),因而该结论说明,离解能的大小与配位键的长短密切相关,其键长越短,离解能越高.另外,表2中的计算结果还显示,离解能随着相对介电常数或基组的增加而减小;尤其是施加扩散函数的计算基组,计算出的离解能随介电常数的增加减少较快.对模型M2,我们采用B3LYP/6-311+G(2d,2p)方法在介质气相中的基态计算结果(0.47 eV)与文献中的理论计算结果(0.49 eV)[16]相比非常接近,也与文献报道的水分子与金属配位的离解能范围(57-40 kJ·mol-1或0.59-0.41 eV)[23]十分吻合.但在蛋白质模拟环境和介质水中的计算结果(0.23和0.09 eV)与文献值(0.37和0.31 eV)[16]相差较大,其主要原因是我们采用的介质的计算模型不同,说明了理论计算的局限和近似性.对模型M3而言,采用B3LYP/6-311+G(2d,2p)方法在气相中的基态计算结果(0.62 eV)与报道的叶绿素与处于氢键束缚下的甲基配位的结合能(56.42 kJ·mol-1或0.58 eV)的计算结果[13]相一致.
2.3 氧化还原势
氧化还原势U是表征分子失去和得到电子能力的量度,由下式估算[24]:

其中4.43 eV是对标准氢电极电势的估计值.采用理论计算方法研究生物分子氧化还原势的关键步骤之一是其所处介质环境的模拟.
表 3给出采用 B3LYP方法结合6-31G(d)、6-311+G(d)和6-311+G(2d,2p)三种基组计算出的三种分子模型在介质气相(ε=1.0)、模拟蛋白质环境(ε=4.0)和水(ε=78.4)中的氧化还原势.由表3可以看出:对同一分子模型而言,计算基组含有扩散函数时,计算出的氧化还原势增高,在同一基组下,氧化还原势随着相对介电常数的增加而减小.而在相同计算方法和介质中,M3和M2分子模型的氧化还原势均小于M1的氧化还原势.也就是说,水分子配位于叶绿素以及水分子配位后再与天冬酰胺有氢键结合均使得合成分子的氧化还原电势减少.
我们的计算结果与文献中的计算结果[16,22](见表3中斜线后的数据)相比较全部偏小.气相时,我们的计算值更接近叶绿素a的实验值(1.67 eV,气态)[25],最佳值是采用6-311+G(2d,2p)基组计算的1.74 eV,这与我们计算所用的分子模型的原子数比文献中保留的多,更接近实际相吻合.另外,表3的结果同样表明,计算基组越大,计算结果越接近实际.
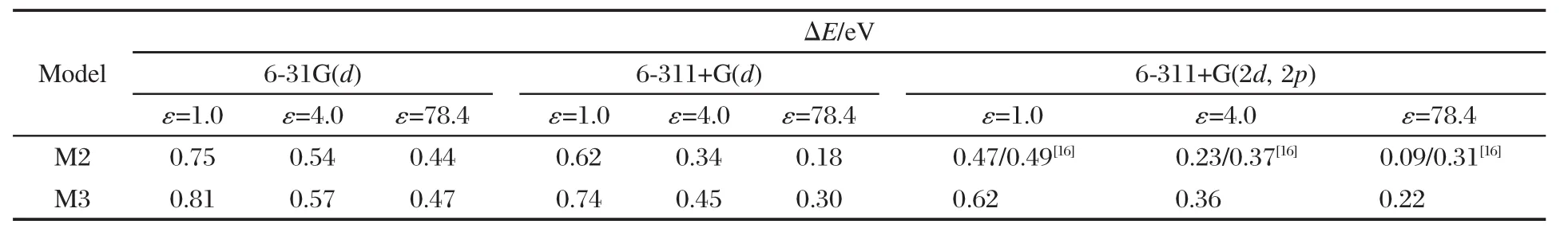
表2 在不同条件下计算出的M2和M3基态离解能(△E)Table 2 Calculated dissociation energy(△E)for molecular models M2 and M3 at neutral state in different conditions
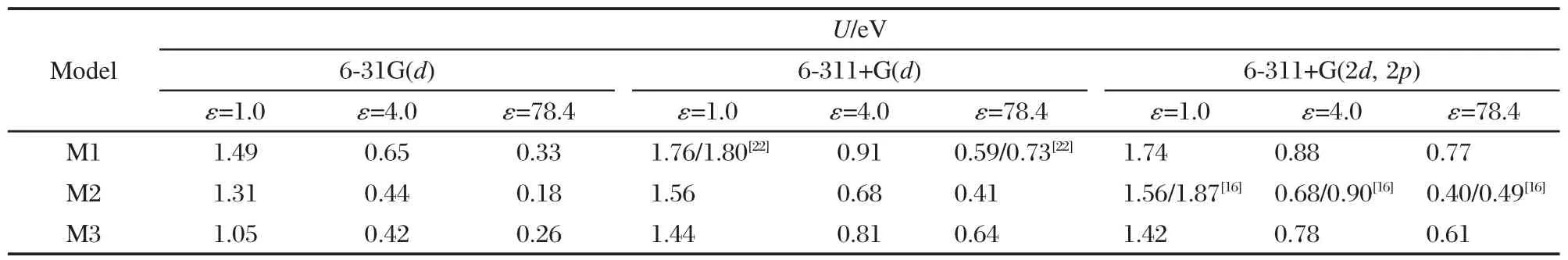
表3 在不同条件下计算出的三种分子模型的氧化还原势(U)Table 3 Calculated redox potential(U)for three molecular models in different conditions
2.4 红外光谱
单叶绿素a分子模型M1含有85个原子,分子模型M2和M3各含有88和105个原子,分别有249、258和309个简谐振动频率,但其中大部分的振动强度非常弱,实验中难以观测到,在此我们仅对特征频率进行讨论.
实验研究指出,单叶绿素a的羧基(C=O)振动峰位位于1650-1740 cm-1之间,而且C133=O和C173=O的振动重叠在1739 cm-1附近,C131=O振动峰位出现在1696 cm-1附近.另外,卟啉环上的C=C耦合振动位于1600 cm-1附近,与镁离子-配体有关的振动位于650-250 cm-1之间,而且振动强度弱,振动耦合复杂,难以指认[2,26].
图3(a)给出理论计算的分子模型M1,M2和M3位于1850-1600 cm-1区间的红外差值光谱.差值光谱由阳离子态的吸收谱减去基态的吸收谱而得.因此,差值光谱中的正值峰位代表阳离子态,负值峰位代表中性态.差值谱给出与阳离子态有关的振动峰位的移动,反映了分子键电子分布的改变.
图3(b)给出理论计算的模型M2位于1850-1550 cm-1区间的基态红外吸收光谱及其同位素15N、2H和13C的标记谱.M1和M3的光谱在此没有给出.值得一提的是,所有理论计算一般高于实验值5%左右[27],由于我们关心的是分子在阳离子态和不同条件下光谱峰位的变化,故没有采用校正因子.
理论计算表明(见图3(a)),1850-1600 cm-1区间含有两个特征差值峰位,分别位于1800和1650 cm-1附近.模型 M1、M2和 M3基态的 1805、1805和1798 cm-1峰位主要归属于C173=O和C133=O的振动耦合,含有C131=O的贡献.处于阳离子态时,该峰位分别上移了 20、18和 22 cm-1,位于 1825、1823和1820 cm-1,但主要归属于C131=O的伸缩振动,掺有C133=O微弱振动的耦合;而1795、1793和1792 cm-1峰位主要归属于M1、M2和M3基态的C131=O和C133=O伸缩振动耦合,当阳离子态形成时,该峰位分别上移 13、15和 16 cm-1,位于1808 cm-1附近,但振动强度减弱,在差值谱中不明显.峰位的上移给出的是微观环境偏极性减低和键长减小的信息[28],反之亦然.基态中1653 cm-1(M1)、1649 cm-1(M2)和1650 cm-1(M3)峰位主要归属于卟啉环所有C=C的振动耦合,与C=O键的情况相反,阳离子态形成时,该峰位分别下移21、17和17 cm-1,位于1632 cm-1/1632 cm-1/1633 cm-1(M1/M2/M3).这两个特征差值峰位与实验中观察到的羧基C=O和卟啉环的C=C特征振动峰位相呼应,但未曾见过C=O键振动耦合的实验报导,因此,该计算结果虽然与文献报导过的计算结果一致[2,17,29],但仍与实验结果存在差异[3,10,30-32].

由图3(b)可看出,上述羧基C=O和卟啉环C=C峰位在模型M2的15N标记的红外吸收谱中没有变化(M1和M3也如此),说明C=O和C=C峰位没有受到氮原子的振动耦合.但对位于1600 cm-1附近的谱带有所变化,说明该振动峰位掺有氮原子的振动耦合,这一结论与现有实验报导[30,32]相一致.13C标记时,C=O键的耦合振动峰位1805 cm-1/1793 cm-1下移了45 cm-1/46 cm-1位于1760 cm-1/1747 cm-1,这与实验中测定的44 cm-1[30,33]相吻合,并且2H标记时下移了4 cm-1/3 cm-1位于1801 cm-1/1790 cm-1,同样与实验中测定的5 cm-1/3 cm-1[30,32-34]相吻合.位于1649 cm-1的卟啉环C=C峰位在13C/2H标记时下移了59 cm-1/12 cm-1而位于 1590 cm-1/1637 cm-1.在模型M2和M3中,该峰位分别下移60 cm-1/12 cm-1和60 cm-1/13 cm-1(13C/2H).
上述分析看出,C=O振动峰位在三种模型中,无论是基态还是阳离子态,计算的频率最多相差7 cm-1/5 cm-1(基态/阳离子态),而卟啉环C=C振动峰位最多变化是4 cm-1/1 cm-1(基态/阳离子态),而且同位素15N/2H/13C标记所引起的这两个峰位的下移量在三种分子模型中也很接近,相差小于3 cm-1,说明配位体和天冬酰胺对所讨论的叶绿素a的红外光谱特征没有显著影响.
3 结 论
对辅助叶绿素a的三种配位分子模型研究表明:水分子配位和配位的水分子受到来自天冬酰胺的氢键束缚均导致叶绿素a的Mg—N键键长增加,以镁离子为中心的键角减小,但对其红外特征光谱的影响不显著;计算出的分子模型M3的离解能均高于M2,且Mg—O配位键的间距小于M2,分子的氧化还原势和配位键离解能随着相对介电常数的增加而减小;采用施加扩散函数的基组计算出的氧化还原势增高;阳离子态的形成引起C=O键的振动频率上移,而C=C键的振动频率下移.同位素标记的计算结果与实验中测定值相吻合.
致谢: 本文所有计算工作均在云南大学高性能计算机中心曙光4000A下完成,在此深表感谢.
1 Jordan,P.;Fromme,P.;Witt,H.T.;Klukas,O.;Saenger,W.;Krauss,N.Nature,2001,411:909
2 Wang,R.;Parameswaran,S.;Hastings,G.Vibrational Spectroscopy,2007,44:357
3 Vernon,L.P.;Seely,G.R.The chlorophylls.New York:Academic Press,1966:187-251
4 Dolphin,D.The porphyrins.New York:Academic Press,1978:430-455
5 Fromme,P.;Jordan,P.;Krauß,N.Biochimica et Biophysica Acta,2001,1507:5
6 Matyushov,D.V.J.Phys.Chem.B,2006,110:10095
7 Webber,A.N.;Lubitz,W.Biochimica et Biophysica Acta,2001,1507:61
8 Krabben,L.;Schlodder,E.;Jordan,R.;Carbonera,D.;Giacometti,G.;Lee,H.;Webber,A.N.;Lubitz,W.Biochemistry,2000,39:13012
9 Ramesh,V.M.;Gibasiewicz,K.;Su,L.;Bingham,S.E.;Webber,A.N.Biochimica et Biophysica Acta,2007,1767:151
10 Holzwarth,A.R.;Muller,M.G.;Niklas,J.;Lubitz,W.Biophysical Journal,2006,90:552
11 Giera,W.;Ramesh,V.M.;Webber,A.N.;Stokkum,I.V.;Grondelle,R.V.;Gibasiewicz,K.Biochimica et Biophysica Acta,2010,1797:106
12 Zhao,G.J.;Han,K.L.Biophysical Journal,2008,94:38
13 Zhao,G.J.;Han,K.L.ChemPhysChem,2008,9:1842
14 Zhao,G.J.;Liu,J.Y.;Zhou,L.C.;Han,K.L.J.Phys.Chem.B,2007,111:8940
15 Zhao,G.J.;Han,K.L.J.Phys.Chem.A,2007,111:9218
16 Heimdal,J.;Jensen,K.P.;Devarajan,A.;Ryde,U.J.Biol.Inorg.Chem.,2007,12:49
17 O′Malley,P.J.Am.Chem.Soc.,2000,122:7798
18 Parameswaran,S.;Wang,R.;Hastings,G.J.Phys.Chem.B,2008,112:14056
19 Frisch,A.;Dennington II,R.D.;Keith,T.A.;Millam,J.GaussView 4.1.2.Wallingford,CT:Gaussian Inc.,2007
20 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03.Revision D.01.Pittsburg,PA:Gaussian Inc.,2004
21 Foresman,J.B.;Frisch,A.Exploring chemistry with electronic s tructuremethods.2nded.Pittsburg,PA:GaussianInc.,1996:1-302
22 Hasegawa,K.;Noguchi,T.Biochemistry,2005,44:8865
23 Shen,Y.;Ryde,U.JournalofInorganicBiochemistry,2004,98:878 24 Reiss,H.;Heller,A.J.Phys.Chem.,1985,89:4207
25 Nakato,Y.;Chiyoda,T.;Tsubomura,H.Bull.Chem.Soc.Jpn.,1974,47:3001
26 Sheer,H.Chlorophylls.Boca Raton:CRC Press,1991:855-902
27 Wheeler,R.A.;Eriksson,L.A.Theoretical biochemistry-processes and properties of biological systems.Amsterdam:Elsevier,2001:655-690
28 Noguchi,T.Photosynth.Res.,2010,104:321
29 Berezin,K.V.;Nechaev,V.V.;Ziganshina,O.D.Journal of Structural Chemistry,2004,45:217
30 Nabedryk,E.;Leonhard,M.;Mantele,W.;Breton,J.Biochemistry,1990,29:3242
31 Breton,J.;Nabedryk,E.;Clerici,A.Biochimica et Biophysica Acta,2001,1507:180
32 Wang,R.;Sivakumar,V.;Johnson,T.W.;Hastings,G.Biophysical Journal,2004,86:1061
33 Schmid,E.D.;Schneider,F.W.;Siebert,F.Spectroscopy of biological molecules-new advance.Chichester:Wiley&Sons 1988:297-300
34 Breton,J.;Nabedryk,E.;Leibl,W.Biochemistry,1999,38:11585
Effect of Water Ligation on the Redox Potential and Infrared Spectra of Chlorophyll-a
HU Qiong WANG Guo-Ying OU Jia-Ming WANG Rui-Li*
(College of Physics and Electronic Information,Yunnan Normal University,Kunming 650092,P.R.China)
O641;O657.3
Received:May 18,2010;Revised:August 4,2010;Published on Web:September 23,2010.
*Corresponding author.Email:ruiliw@yahoo.com;Tel:+86-871-6788724.
The project was supported by the National Natural Science Foundation of China(10764006).
国家自然科学基金(10764006)资助项目
