HPLC-UV-FTICRMS/MSn法分离鉴定葛根素原料药中4种有关物质
2010-09-18王鹏远吴彩胜张金兰
王鹏远,吴彩胜,吴 松,张金兰
(中国医学科学院,北京协和医学院,药物研究所,北京 100050)
HPLC-UV-FTICRMS/MSn法分离鉴定葛根素原料药中4种有关物质
王鹏远,吴彩胜,吴 松,张金兰
(中国医学科学院,北京协和医学院,药物研究所,北京 100050)
采用高效液相色谱-紫外检测器-傅里叶变换离子回旋共振质谱(HPLC-UV-FTICRMS/MSn)技术获得了葛根素及其有关物质的 MSn和 HRMS数据,分析其可能的裂解机制,推断有关物质的结构,并依据HRMS数据确证了有关物质及其部分碎片的分子组成,最终鉴定葛根素原料药中4种有关物质,其准分子离子峰[M+H]+分别为 m/z433.112 95、433.112 92、417.118 01和417.118 01,通过质谱裂解机制研究以及与文献数据的对比,推测它们的结构分别为3’-羟基葛根素、染料木素-8-C-葡萄糖苷、新葛根素B和新葛根素A。
葛根素;电喷雾电离(ESI);傅里叶变换离子回旋共振质谱(FTICR-MS);结构鉴定
近年来,利用高效液相色谱-紫外检测器-傅里叶变换离子回旋共振质谱(HPLC-UV-FTICRMS/MSn)技术鉴定原料药及制剂中的有关物质结构[1-4],已成为一种快速、准确、灵敏的方法,为药物的生产、贮存、运输及临床应用过程中的质量安全控制提供了高效便捷的手段。由于原料药是药物制剂产品的起始材料和主要成分,对其进行有关物质的检测鉴定直接关系到后续产品的质量安全,因此建立快速、准确的分析方法来鉴定原料药中有关物质的结构具有重要意义。
葛根素(puerarin)是由豆科植物野葛 Pueraria lobata(Willd.)Ohwi的干燥根中提取分离得到的 8-β-D-葡萄吡喃糖-4′,7-二羟基异黄酮[5],结构示于图1。葛根素具有扩张血管、促进血液循环、改善血液流变性等作用,在治疗心脑血管疾病方面具有广泛应用[6-7]。当前,有关葛根素及其制剂引发药物不良反应的病例屡有报道[8],原因可能与其中的不明杂质有关。
线性离子阱(L TQ)的灵敏度比三维离子阱(LCQ)提高了1个数量级,可以获得更高的采集速度、分辨率及高质量的MSn谱图。傅里叶变换离子回旋共振质谱(FTICR-MS)具有灵敏度高(10-18mol),分辨率高(在500 000半峰宽以上),外标法可获得高质量精度(1~2μg·g-1),是目前世界上测量准确度和分辨率最高的质谱。线性离子阱-傅里叶变换离子回旋共振质谱(L TQ-FTICR-MS)综合L TQ的稳定性、多样性、MSn能力和 FT的高分辨、精确质量测定能力[9],可以在获得微量组分的多级质谱碎片的同时对其进行分子组成的测定,所有工作在一次分析中完成,为快速准确鉴定物质结构提供了技术手段。本工作拟采用 HPLC-UV-FTICRMS/MSn方法快速分离鉴定葛根素原料药中有关物质,通过MSn数据推断其结构,并使用高分辨质谱数据确证其分子组成。
1 实验部分
1.1 仪器与材料
Surveyor LC Plus高效液相系统:美国Finnigan公司产品,配有MS泵、自动进样器和光电二极管阵列检测器(PDA);线性离子阱-傅里叶变换离子回旋共振质谱仪:美国 Finnigan公司产品,配有电喷雾离子源(ESI)及 Tune Plus工作软件和Xcalibur 2.0.7数据处理系统。

图1 葛根素及其有关物质的结构和相对分子质量Fig.1 The structures and relative molecular weights of puerarin and its related impurities
甲醇(农残级)、乙腈(农残级)、乙酸(分析级):美国 Mallinckrodt Baker公司产品;纯净水:杭州娃哈哈公司产品,使用前经0.45μm微孔滤膜过滤;葛根素对照品(批号:110752-200511):中国药品生物制品检定所提供;两批葛根素原料药,由中国医学科学院药物研究所吴松研究员提供。
1.2 色谱条件
Agilent公司 ZORBAX Eclipse XDB C18色谱柱(250 mm ×4.6 mm ×5μm);流动相:水(A,用乙酸调p H为3.0)和乙腈(B);梯度洗脱,洗脱程序:0~5min,5%~14%B;5~10 min,14%B;10~11 min,14%~18%B;11~20 min,18%~22%B;流速1 mL·min-1;柱温30℃;进样量10μL 或20μL;检测波长250 nm;分流比为7∶3。
1.3 质谱条件
ESI离子源,采用正离子检测模式;喷雾电压3.5 kV;毛细管温度275℃;鞘气(N2)流速35 arb;辅助气(N2)流速10 arb;扫尾气(N2)流速3 arb;管状透镜:110 V;碰撞能量35 V;扫描范围100~800 u;扫描方式:L TQ采用全扫描方式 MS5扫描,且MS2采用数据相关扫描模式对 m/z433、417进行碰撞诱导解离(CID)全扫描,MS3~MS5则依次对上一级谱图中最强离子峰进行CID全扫描;FT采用全扫描方式 MS2扫描,且MS2采用Data Dependent Scan模式对m/z433、417进行CID全扫描。
1.4 样品处理
精密称取0.1 mg葛根素对照品(puerarin Std)及 2 mg第一批葛根素原料药(puerarinⅠ)、2 mg第二批葛根素原料药(puerarinⅡ),分别用1 mL甲醇溶解,涡旋混匀,过0.45μm滤膜,待测。
2 结果与讨论
2.1 HPLC分析
将1.4项下的样品puerarin Std、puerarinⅠ和puerarinⅡ分别注入Surveyor LC Plus高效液相色谱仪,按1.2项的色谱条件测定。结果显示,葛根素原料药中除主药外,还存在4种有关物质,按出峰顺序将其依次记作 S1、S2、S3和S4,紫外图谱显示 S1和 S2有明显区别,葛根素、S3和S4几乎没有区别,后三者可能为同一母核,示于图2。值得注意的是,葛根素化学对照品中也含有这4种有关物质,只是含量低于原料药,说明这些有关物质是葛根素中常见的较难除去的杂质,提示其可能来源于药材的提取分离过程中,由葛根素原药材引入。
2.2 HPLC-ESI-MSn分析
按1.2、1.3项的分析条件,在LC流出组分进入ESI源之前,采用三通进行分流,使得每分钟仅有300μL组分进入质谱仪,以保证充分的离子化效果。其中puerarin Std是以0.1 g·L-1的浓度进样10μL,由LC-UV图得知葛根素的保留时间约为9.5 min。为了提高原料药中有关物质的检测灵敏度,将puerarinⅠ、Ⅱ以2 g·L-1的浓度进样20μL,并在 8.5~11.5 min区段把LC流出的高浓度葛根素组分切换至Waste阀,以防其污染质量分析器。
葛根素原料药(第 Ⅰ批)的总离子流图(TIC)示于图 3,可以明显观察到 S1、S2、S3和S4的[M+H]+峰依次为 m/z433、433、417 和417,是2对同分异构体,葛根素的[M+H]+峰也是 m/z417,S1、S2的[M+H]+峰比其多16 u,可能为葛根素的羟基化产物。通过分别对m/z433和417的MS2图谱进行中性丢失碎片为120 u的提取,可以观察到4种有关物质都有显著的[M+H-120]+峰,提示它们都为碳苷类化合物[3-4,10],示于图4。葛根素原料药(第Ⅰ批)的 HPLC-MSn数据列于表1,实验获得的葛根素MS1~MS5数据均与文献[10]一致,具有相同的特征碎片离子 m/z399、381、363、351、321、297和267,裂解行为相同,其机制不再赘述。

图2 葛根素对照品(a)和原料药(b,第Ⅰ批)的 HPLC-UV图谱Fig.2 HPLC-UVchromatogram of puerarin standard(a)and bulk drug(b,batch No.1)
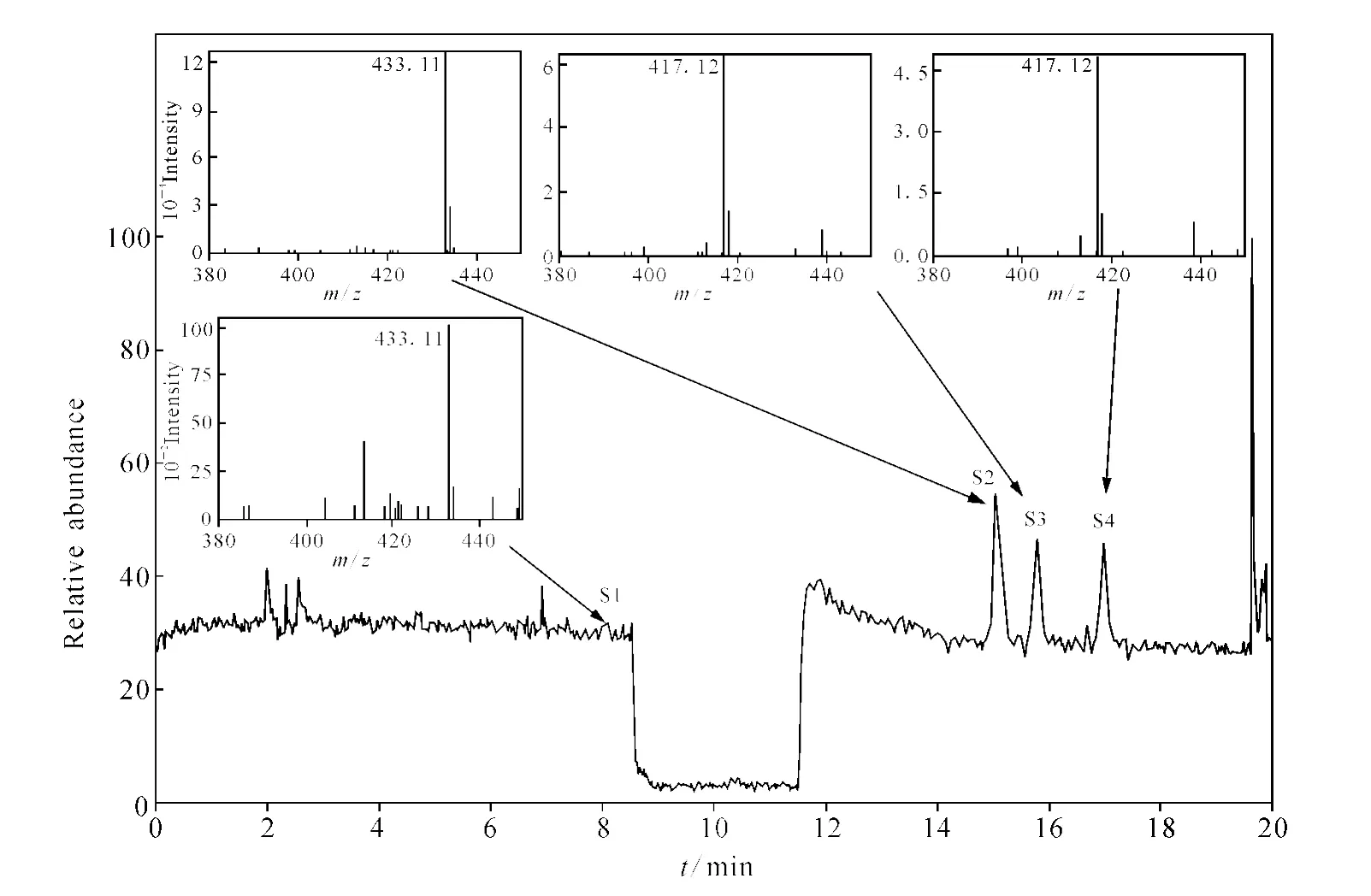
图3 葛根素原料药(第Ⅰ批)的LTQ-MS总离子流图(TIC)Fig.3 LTQ-MS total ion current chromatogram of puerarin bulk drug(batch No.1)
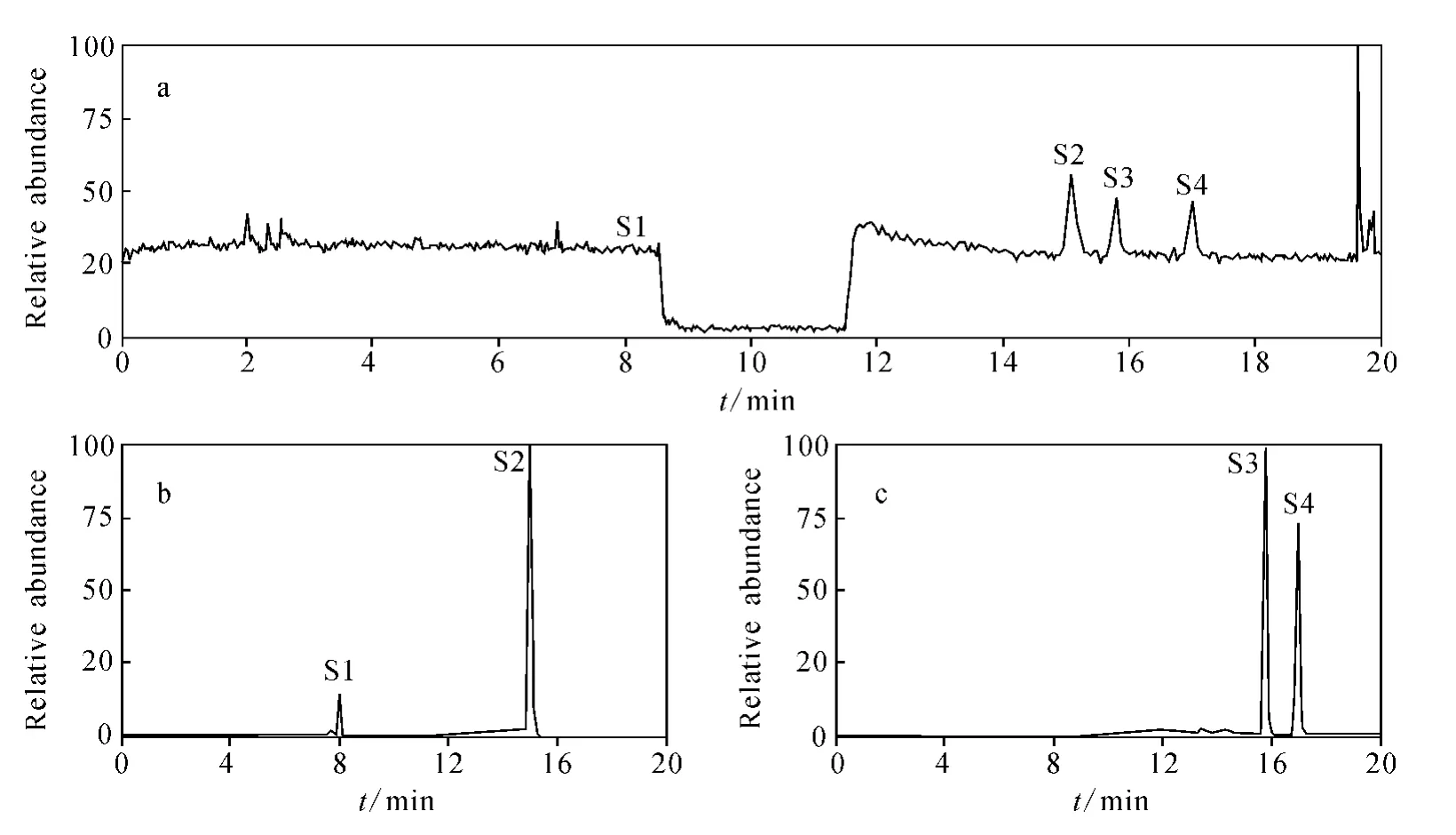
图4 葛根素原料药(第Ⅰ批)的 TIC图(a)及分别对 m/z433(b)和m/z417(c)的MS2图谱进行中性丢失碎片为120 u的离子流提取图谱Fig.4 The TIC chromatogram(a)of puerarin bulk drug(batch No.1)and the extracted ion current chromatogram of neutral ioss fragment 120 u form/z433(b)andm/z417(c)
S1和 S2的MS2、MS3和 MS4谱图基本相同,主要都在糖环上发生脱 H2O和CH2O反应 ,形成特征碎片离子 m/z415、397、379、367、351、337、319、313、309、295、291、281、269、263和253,其裂解机制示于图 5。但是,在 MS4图谱中,S1出现特有的碎片离子 m/z203和161,且MS5图谱中 S1出现特有的碎片离子 m/z 175,S2出现特有的碎片离子m/z191和163,都为异黄酮母核经过C环的RDA裂解得到的A环残基,并进一步脱C2H2O和CO产生,其裂解机制示于图6。其中S2的碎片离子m/z191比S1的m/z175刚好多出16 u,提示S2的羟基化位置在A 环,而 S1在B环,且 S1中m/z337离子裂解产生碎片离子m/z203和161,进一步证明了S1的羟基化位置在B环,再根据二者的UV图特征和在反相色谱中的保留行为特征,S1在231 nm和288 nm处有最大吸收,而S2只在261 nm处有最大吸收,且S1比S2先被洗脱出来,与文献数据一致[3-4,11-12],即可确定 S1为3’-羟基葛根素,S2为染料木素-8-C-葡萄糖苷。
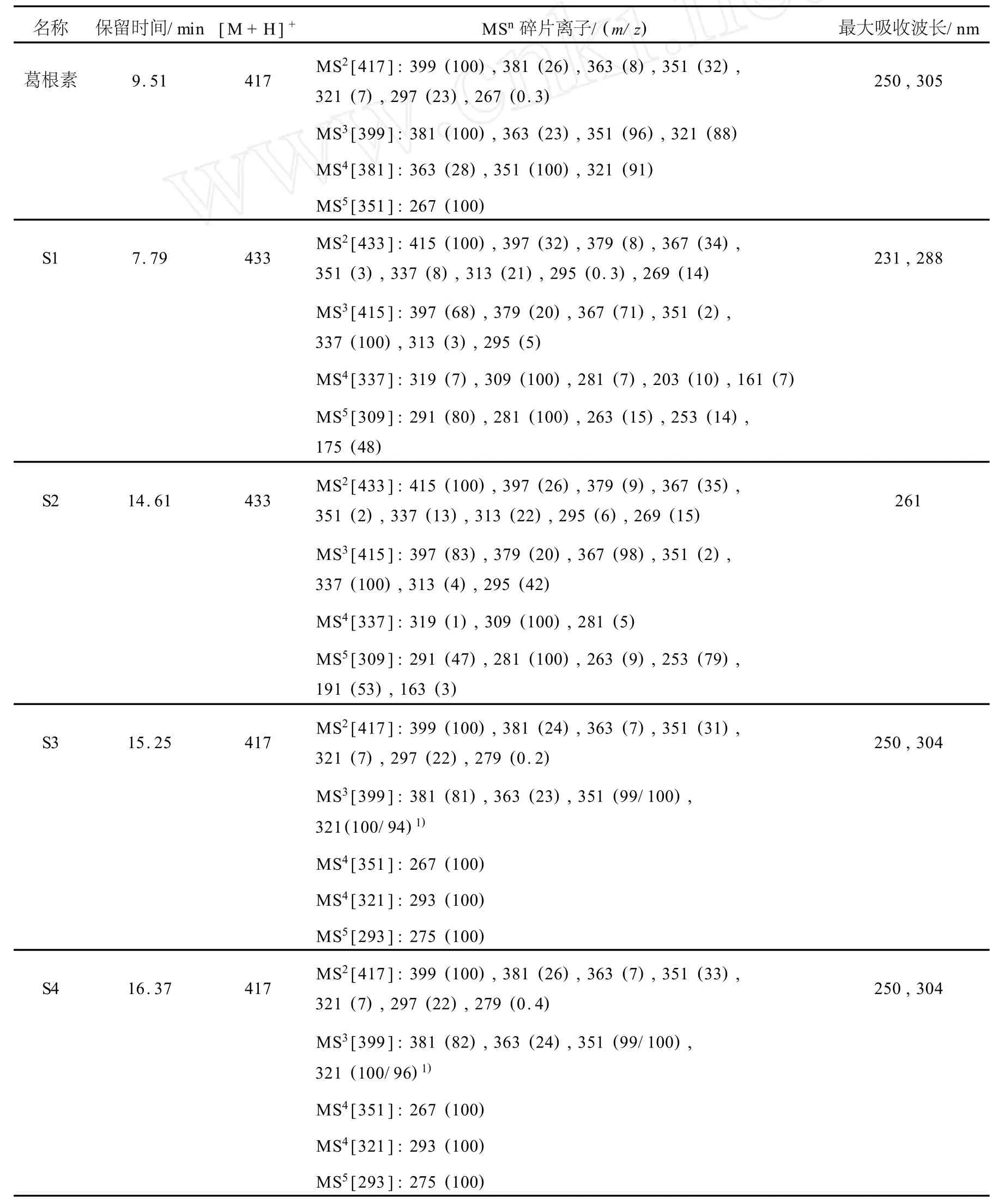
表1 葛根素原料药(第Ⅰ批)的 HPLC-MSn数据Table 1 HPLC-MSnanalysis of puerarin bulk drug(Batch No.1)

图5 有关物质S1、S2相同部分的裂解机制Fig.5 The same part of ESI-MSnfragment mechanism of related impurities S1,S2
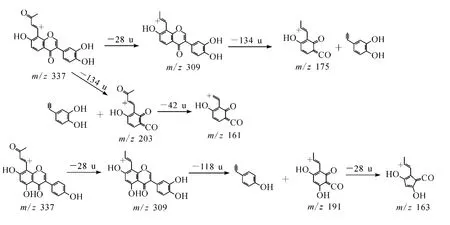
图6 有关物质S1、S2特征部分的裂解机制Fig.6 The characteristic part of ESI-MSnfragment mechanism of related impurities S1,S2
S3、S4与葛根素是同分异构体,它们的MSn数据基本相同,其裂解机制示于图7,且UV图谱基本相同,说明三者可能是相同母核。根据已知的国内外文献[4,13],目前从葛根中鉴定出来的相对分子质量为416 u的同分异构体共有4种,分别是葛根素、新葛根素、新葛根素A,新葛根素B,其中新葛根素在文献中没有可参考的UV数据,而且其在反相色谱中紧接着主峰葛根素洗脱出,而新葛根素 A和新葛根素B与 S3、S4的UV特征吸收峰以及在反相色谱中的保留行为特征符合,它们都在250、304或305 nm处有最大吸收,且新葛根素B先于新葛根素A被洗脱出,故确定S3为新葛根素B,S4为新葛根素A。
2.3 ESI-FTICR-MS2分析
FTICR-MS相当于L TQ的另一台高分辨、高精度、高灵敏度的检测器,将L TQ产生的丰富的MSn碎片离子进行精确分子质量测定,进而确定其分子组成。葛根素对照品和原料药的高分辨质谱图示于图8。从图中可知,葛根素、S1、S2、S3和 S4的准分子离子峰[M+H]+分别为 m/z417.117 95、433.112 95、433.112 92、417.118 01和417.118 01,经质谱精密质量检索得其[M+H]+的元素组成分别为C21H21O9(计算值为 417.118 01,误差为 -0.14×10-6)、C21H21O10(计算值为433.112 92,误差为0.06×10-6)、C21H21O10(计算值为 433.112 92,误差为0)、C21H21O9(计算值为 417.118 01,误差为 0)和C21H21O9(计算值为417.118 01,误差为0),非常准确的得出葛根素及其有关物质的分子组成。根据高分辨二级质谱数据可以确定各主要碎片的分子组成,进而得出丢失碎片的分子组成,为依据L TQ-MSn数据推断化合物的质谱裂解机制提供了准确可靠的信息。葛根素原料药(第Ⅰ批)的 ESI-FTICR-MSn数据及各碎片的理论计算值和误差列于表2,有关物质S1的浓度很低,没有获得有效的二级高分辨质谱数据。经计算,所有碎片误差绝对值的平均值仅为0.52×10-6,显示出了 FTICR-MS测定精确分子质量的独特能力。
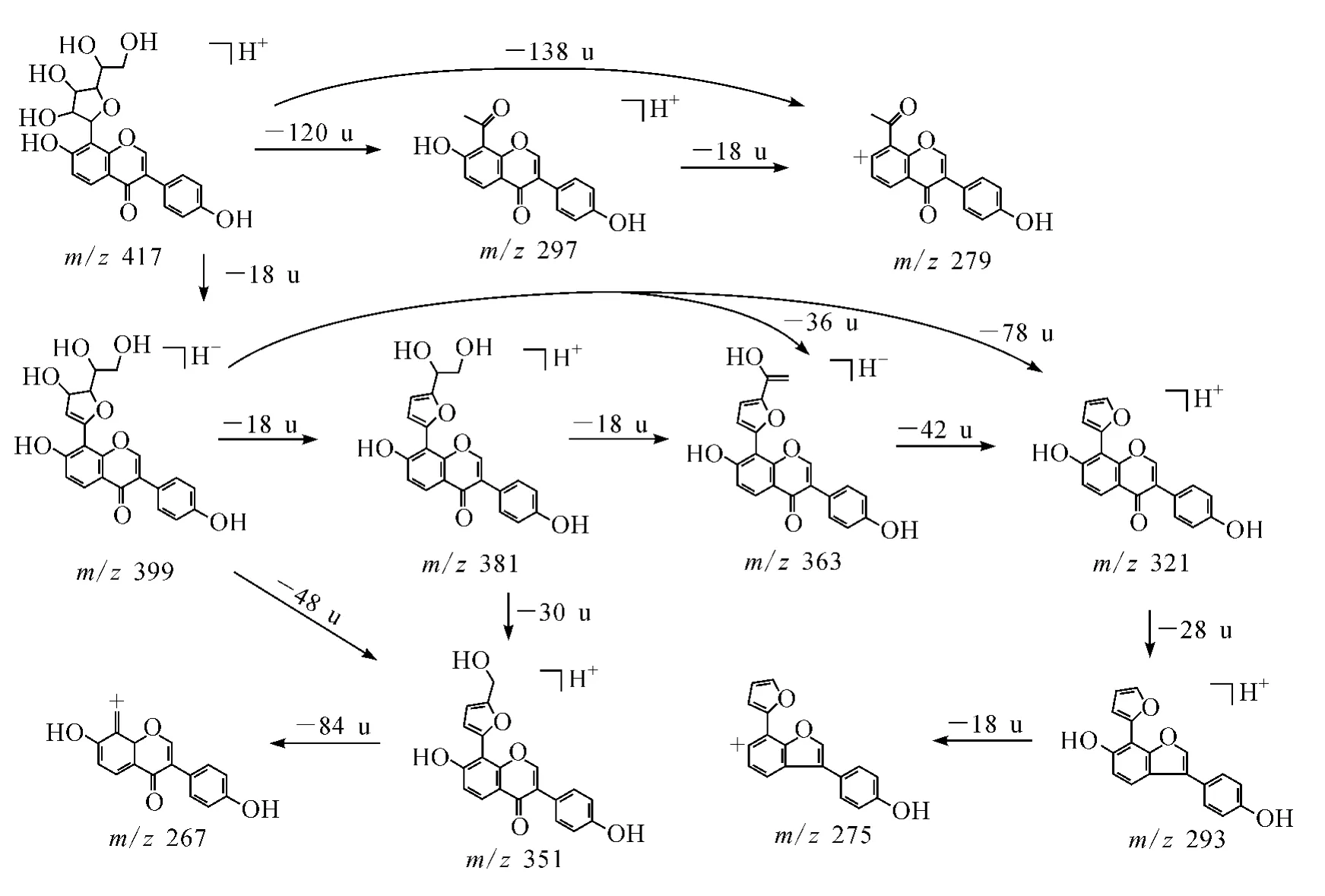
图7 有关物质S3、S4(二者相同)的裂解机制Fig.7 The ESI-MS fragment mechanism of related impurities S3,S4(the two are the same)
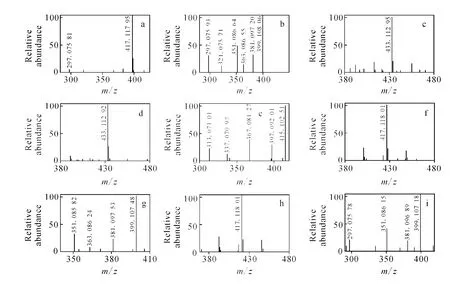
图8 a.葛根素的一级高分辨质谱图;b.葛根素m/z417离子的二级高分辨质谱图;c.S1的一级高分辨质谱图;d.S2的一级高分辨质谱图;e.S2的m/z433离子的二级高分辨质谱图;f.S3的一级高分辨质谱图;g.S3的m/z417离子的二级高分辨质谱图;h.S4的一级高分辨质谱图;i.S4的m/z417离子的二级高分辨质谱图Fig.8 a.HR-MS1spectrum of puerarin;b.HR-MS2spectrum of puerarin[M+H]+m/z417;c.HR-MS1spectrum of S1;d.HR-MS1spectrum of S2;e.HR-MS1spectrum of S2[M+H]+m/z433;f.HR-MS1spectrum of S3;g.HR-MS1spectrum of S3[M+H]+m/z417;h.HR-MS1spectrum of S4;i.HR-MS1spectrum of S4[M+H]+m/z417
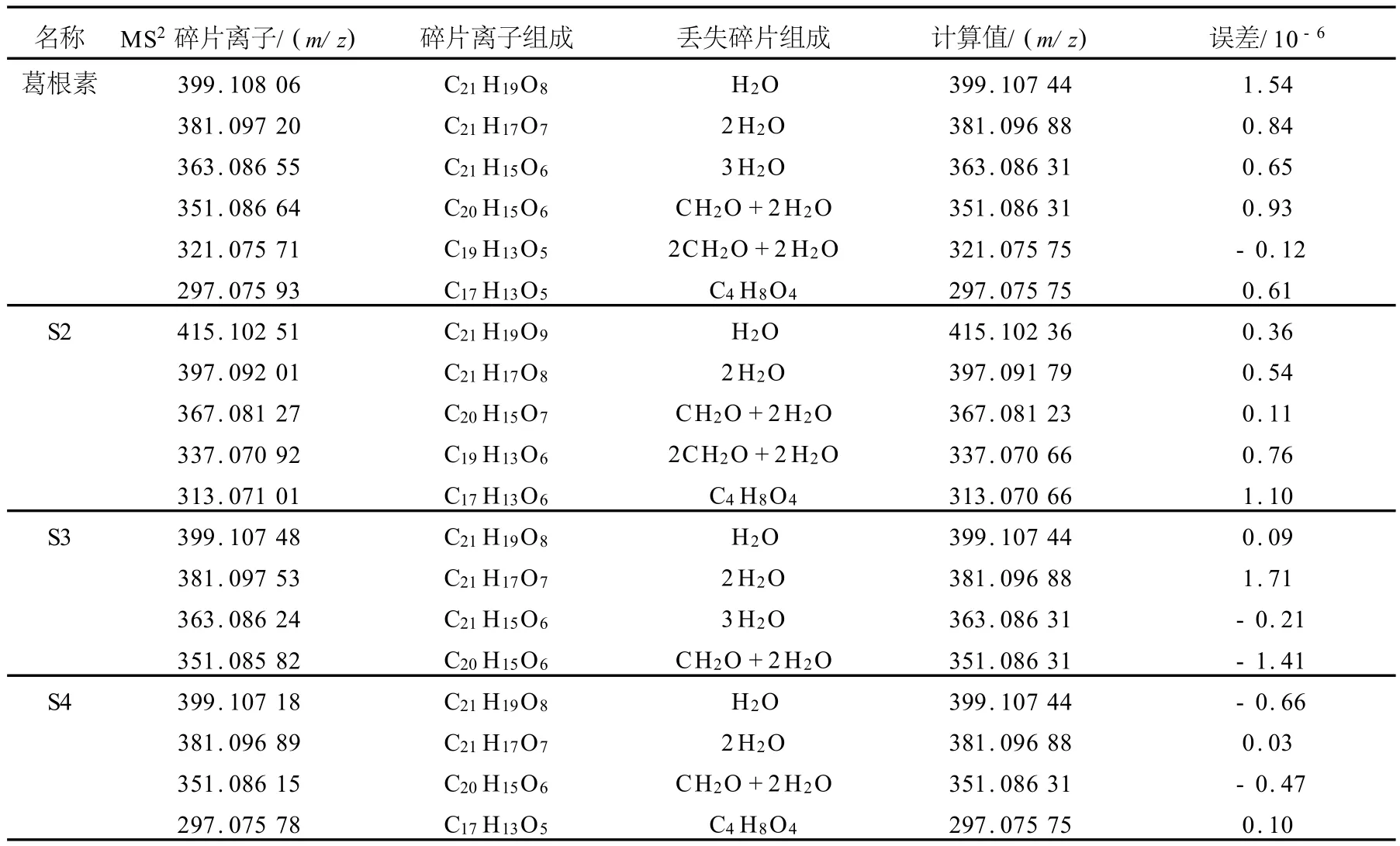
表2 葛根素原料药(第Ⅰ批)的 ESI-FTICR-MSn数据Table 2 ESI-FTICR-MSnanalysis of puerarin bulk drug(batch No.1)
3 结 论
有关葛根素原料药及其注射剂中有关物质的分离分析已有文献报道,但目前还没有对葛根素原料药中4种有关物质同时进行快速准确的鉴定,而且也没有文献报道过葛根素及其4种有关物质的高分辨质谱数据以及3’-羟基葛根素、染料木素-8-C-葡萄糖苷苷元部分的质谱裂解数据和分析。张璐等[3]曾报道采用LC/MS技术对葛根素原料药及注射剂中的有关物质结构进行初步鉴定,在9批原料药中有8批检出了4种有关物质,但仅初步鉴定出2种物质结构,MSn只做到异黄酮苷的糖环裂解部分,没有苷元的裂解数据和分析,不能通过MSn数据确定苷元羟基化位置,而且LC分析时间长达60 min,本方法仅用20 min。Zhang等[4]报道了采用LC/MS技术对葛根素注射液中有关物质的分析,虽然在35 min内分离出9种有关物质,并且根据文献数据鉴定出其中8种有关物质的结构(包含本文中鉴定出的4种),但没有高分辨质谱数据支持,而且没有对葛根素原料药进行分析。本研究根据L TQ-MS获得的丰富的MSn数据推断出4种有关物质的质谱裂解机制,并且根据 FTICRMS获得的高分辨MSn数据为上述结构确定提供准确依据,建立了快速、准确的鉴定葛根素原料药中4种有关物质的方法,其结构确定为3’-羟基葛根素、染料木素-8-C-葡萄糖苷、新葛根素B和新葛根素A,对于明确葛根素原料药的成分组成和质量控制具有重要意义。
[1] 覃洁萍,邓家刚,冯钰锜,等.有机质谱分析在芒果苷原料药微量杂质结构鉴定中的应用[J].质谱学报,2008,29(4):218-214.
[2] ZHOU H,ZHENG Z G,WU S H,et al.Separation and characterization of clindamycin and related impurities in bulk drug by high-performance liquid chromatography-electrospray tandem mass spectrometry[J].J Pharm Biomed Anal,2006,41:1 116-1 123.
[3] 张 璐,陈民辉,蔡美明,等.HPLC和LC-MSn分析葛根素及其注射剂中的有关物质[J].中国生化药物杂志,2008,29(6):361-365.
[4] ZHANG H J,YANG X P.Profiling and quantification of isoflavone-c-glycosides impurities in puerarin injection by liquid chromatography coupled to esi-ion trap mass spectrometry[J].J Pharm Biomed Anal,2009(49):843-847.
[5] 国家药典委员会.中国药典:二部[M].北京:化学工业出版社,2005.
[6] 陈荔坦,陈树和,刘焱文.葛根资源、化学成分和药理作用研究概况[J].时珍国医国药,2006,17(11):2 305-2 306.
[7] 叶江枫,杜志强,赵中苏,等.葛根素对兔肺心病模型肺动脉高压的影响[J].中国中医药科技,2001,8(5):321.
[8] 蔡 泳.5种常用中药注射液的不良反应分析[J].医药导报,2006,25(4):370-371.
[9] 卞利萍,张洪杰.线性离子阱-傅里叶变换离子回旋共振质谱的结构原理及在蛋白质组学研究中的应用[J].质谱学报,2005,26(4):247-253.
[10] 李宇航,戴学海,李晓蓉,等.葛根素的电喷雾电离裂解规律解析[J].质谱学报,2007,28(4):224-227.
[11] KINGJ E,FURUSAWA J I,BABA J,et al.Studies on the constitunets of pueraria lobata Ⅲisoflavonoids and related compounds in the roots and voluble stem[J].Chem Pharm Bull,1987,35(12):4 846-4 850.
[12] OHSHIMA Y,OKU YAMA T,TAKAHASHI K,et al.Isolation and high performance liquid chromatograhy(HPLC)of isoflavonoids from the pueraria root[J].Planta Medica,1988,54:250-254.
[13] ZHANG H J,YANG X P,WANG K W.Two new c-glucofuranosyl isoflavones in puerarin injection[J].Chinese Chem Lett,2009,20:720-723.
Separation and Characterization of 4 Related Impurities in Puerarin Bulk Drug by HPLC-UV-FTICRMS/MSn
WANG Peng-yuan,WU Cai-sheng,WU Song,ZHANGJin-lan
(Institute of Materia Medica,Chinese Academy of Medical Sciences and Peking Union Medical College,Beijing100050,China)
The molecular structures and mechanism for the fragmentations of the related impurities were proposed by the MSnand HRMS results obtained by high performance liquid chromatography-ultraviolet detector-fourier transform ion cyclotron resonace mass spectrometry(HPLC-UV-FTICRMS/MSn).The molecular compositions of the related impurities and some of the fragmentations were confirmed by the HRMS datas.Four related impurities in puerarin bulk drug were separated.Their[M+H]+ion peaks arem/z433.112 95,433.112 92,417.118 01 and 417.118 01,respectively.They are identified as 3’-hydoxypuerarin,genistin-8-C-glucoside,neopuerarin B and neopuerarin A on the basis of fragmentation mechanism and reference data.
puerarin;electrospray ionization(ESI);fourier transform ion cyclotron resonance mass spectrometry(FTICR-MS);structure characterization
O 657.63
A
1004-2997(2010)03-0143-09
2009-10-22;
2010-01-28
卫生行业科研专项项目(200802038)资助
王鹏远(1985~),男,山西芮城人,硕士研究生,从事药物分析研究。E-mail:wpy@imm.ac.cn
张金兰(1969~),女,内蒙古呼和浩特人,研究员,从事药物分析研究。E-mail:zhjl@imm.ac.cn
