大气颗粒物中三类有机组分的萃取分离净化和GC/MS测定
2010-09-01段凤魁贺克斌
段凤魁,贺克斌
(环境模拟与污染控制国家重点联合实验室,清华大学环境科学与工程系,北京 100084)
大气颗粒物中三类有机组分的萃取分离净化和GC/MS测定
段凤魁,贺克斌
(环境模拟与污染控制国家重点联合实验室,清华大学环境科学与工程系,北京 100084)
大气颗粒物中有机组分污染特征的研究,对我国城市空气质量监控与治理有重要意义。应用中流量PM2.5采样器采集2003年9月~2004年7月北京市大气颗粒物样品,建立了同时测定PM2.5中三类有机组分的实验方法。采用14%BF3/CH3OH溶液为有机酸衍生化试剂,并确定有机酸最佳衍生化反应温度和时间分别为40℃、45 min;采用自填SPE硅胶柱,实现了正构烷烃、多环芳烃和有机酸酯的完全分离,三类组分回收率都在70%~130%之间。采用GC/MS法定量检出76种化合物,包括C10~C34之间的25种正构烷烃,16种美国EPA优控PAHs,35种有机酸,这些有机酸包括diC2~diC11之间的10种二元酸、C10~C32之间的23种饱和脂肪酸以及C18:1、C18:22种不饱和酸。
大气颗粒物;正构烷烃;多环芳烃;有机酸;GC/MS;衍生化
大气颗粒物中有机组分种类繁多,来源复杂。其中,正构烷烃是一类重要的痕量有机污染物,一般被视为地球化学的生物标志物,根据其污染特征、化学组成可以识别气溶胶的化石燃料来源或植物来源。多环芳烃是被发现和研究较早的一类化学致癌物之一,已经被世界上多数国家列为环境监测的重要内容。美国环保总署确定了16种优控PAHs,我国则将苯并[a]芘等7种多环芳烃列为优先检测黑名单。相对于多环芳烃,脂肪酸虽然生物危害较小,却因为其含量高、吸湿性及与二次有机气溶胶形成有关等特性受到越来越多的关注[1-2]。
大气颗粒物中有机组分污染特征的研究,对城市空气质量监控与治理有重要意义。目前,国内对大气颗粒物中正构烷烃、多环芳烃的研究报道较多,而对于有机酸类,因极性特征难以直接用GC或GC/MS测定,需要通过化学反应使之转化成易挥发的酯类,进而用色谱测定(该化学反应过程称为衍生化[3]),一般比较繁琐。对于细颗粒物(PM2.5)而言,我国已有的报道往往侧重于某一类有机组分[4],而同时测定 PM2.5中几类有机组分的研究还相对较少。
本研究以正构烷烃,多环芳烃和有机酸三类有机污染物为研究对象,在探索超声萃取、固相萃取净化/分离、衍生化方法在内的样品预处理方法以及GC/MS分析方法的基础上,建立同时测定PM2.5中三类有机组分的实验方法。
1 实验部分
1.1 大气颗粒物样品采集
本研究在北京海淀区清华园设采样点进行PM2.5采样,关于采样点的具体描述见文献[5]。研究发现,清华园采样点PM2.5的质量浓度及其化学组分质量浓度与城区代表监测点车公庄的相应浓度处于同一污染水平[5],因此认为清华园采样点可以作为北京城区的代表性观测点。
本研究应用北京地质仪器厂生产的中流量PM2.5单通道采样器进行 PM2.5短期监测。该仪器切割头按照惯性碰撞原理设计,切割粒径为(2.5±0.2)μm,颗粒物采集误差≤±5%。采样时段为2003年9月~2004年7月,每月至少采集4个气溶胶样品,每个样品采集24~48 h,采样流量77.56 L·min-1。中流量采样器的有关原理与小流量采样器的采样结果比对,以及采样过程的质量控制与保证详见文献[6]。
采样器被放置距地面约3~4 m的平台上,气流入口距地面约4.5~5.5 m。用于进行有机组分分析的样品所用的滤膜均为石英纤维滤膜(#2500QAT-UP),直径为90 mm。使用前于500℃灼烧2 h,以除去可能吸附的有机污染物。所采集的气溶胶样品均用铝箔密封于-4℃储存,直到分析测试。
1.2 有机组分的分析方法
1.2.1 标准样品与化学试剂 本研究采用的标准样品包括以下几种:1)16种正构烷烃标样,包括 C15~C28烷、C30烷和 C32烷(99%,Sigma-Aldrich公司产品)。使用时分别称取一定质量标样,溶于正己烷中,配成母液备用。2)16种美国EPA优控多环芳烃混合标样(Chem Service公司产品),使用时加以稀释。3)8种一元脂肪酸甲酯(C10~C24中的偶数碳脂肪酸甲酯,99%, Chem Service公司产品);5种二元脂肪酸甲酯(diC2~diC6,99%,Chem Service公司产品);脂肪酸替代物硬脂酸-d35(Chem Service公司产品)。4)内标六甲苯(99%,Supelco公司产品)。5)PAHs替代物蒽-d10(Sigma-Aldrich公司产品)。
所用化学试剂甲醇、正己烷、二氯甲烷、丙酮等均为色谱纯,购自Dikma公司;衍生化试剂为14%BF3/CH3OH溶液,购自Dikma公司;无水Na2SO4,分析纯,北京化学试剂厂产品,600℃下活化8 h;硅胶,100~200目,Supelco公司产品,140℃下活化12 h。
1.2.2 仪器与设备 样品前处理所用仪器包括超声波清洗器、漩涡混合器、高纯水仪、20孔固相萃取装置及固相萃取柱:Varian公司产品;分析仪器为 Theremo Finnign公司生产的气相色谱-质谱联用(GC/MS)仪(Trace GC/DSQ),配有石英毛细管柱(DM-5MS,30 m×0.25μm× 0.25 mm)。
实验所用全部玻璃仪器均用洗涤剂在自来水中浸泡并超声清洗,然后依次用自来水、去离子水彻底冲洗干净,再用丙酮淋洗,最后在马弗炉中400℃下烘烧5 h,自然冷却后备用。
1.2.3 样品处理 在建立样品前处理方法的基础上,剪取一定量PM2.5样品,放入锥形瓶中,加入C18酸-d35和蒽-d10等代用标准,加入萃取溶剂后密封,进行超声萃取,每次30 min。用20mL C6H14与CH2Cl2分别萃取2、3次,提取非极性、弱极性、中等极性/极性组分,合并萃取液,旋转蒸发浓缩至3 mL左右,进一步用高纯N2浓缩至近干。加入 14%BF3/CH3OH衍生化试剂,于40℃水浴下反应45 min。反应完毕,加入1 mL饱和Na2SO4溶液,用正己烷萃取3次,合并萃取液后浓缩至约1 mL,然后用固相萃取柱进行净化和分离,将有机组分分成正构烷烃、多环芳烃和有机酸酯三大类。
1.2.4 仪器分析条件 使用 Finnigan GC Trace型 GC/MS联用仪分析三类有机组分。升温程序:初温50℃,保持2 min,然后以10℃·min-1升至280℃后保持10 min,进样口温度250℃,检测器温度250℃;进样量均为1 μL,不分流进样;载气为高纯 He气(99.999%)。采用全扫描模式对化合物进行定性,扫描质量范围 m/z100~650。在 GC/MS全扫描基础上,结合谱图库检索与标准物质保留时间对目标化合物进行定性,然后采用选择离子模式(SIM),根据标准物质的工作曲线进行定量(外标法)。
16种正构烷烃、16种多环芳烃标样和13种有机酸酯标样的色谱图示于图1。正构烷烃标样谱图中1~16#峰依次为 C15~C28、C30、C32;多环芳烃1~16#峰依次为萘Na、苊Acy、二氢苊Ace、芴Fl、菲Ph、蒽An、荧蒽Flu、芘Pyr、苯并(a)蒽BaA、屈Chr、苯并(b)荧蒽BbF、苯并(k)荧蒽BkF、苯并(a)芘BaP、茚(123cd)并芘InP、二苯并(ah)蒽DbA和苯并(ghi)苝BghiP;有机酸酯1~13#峰依次为diC2~diC6二元酸酯、C10、C12、C14、C16、C18、C20、C22、C24脂肪酸甲酯。
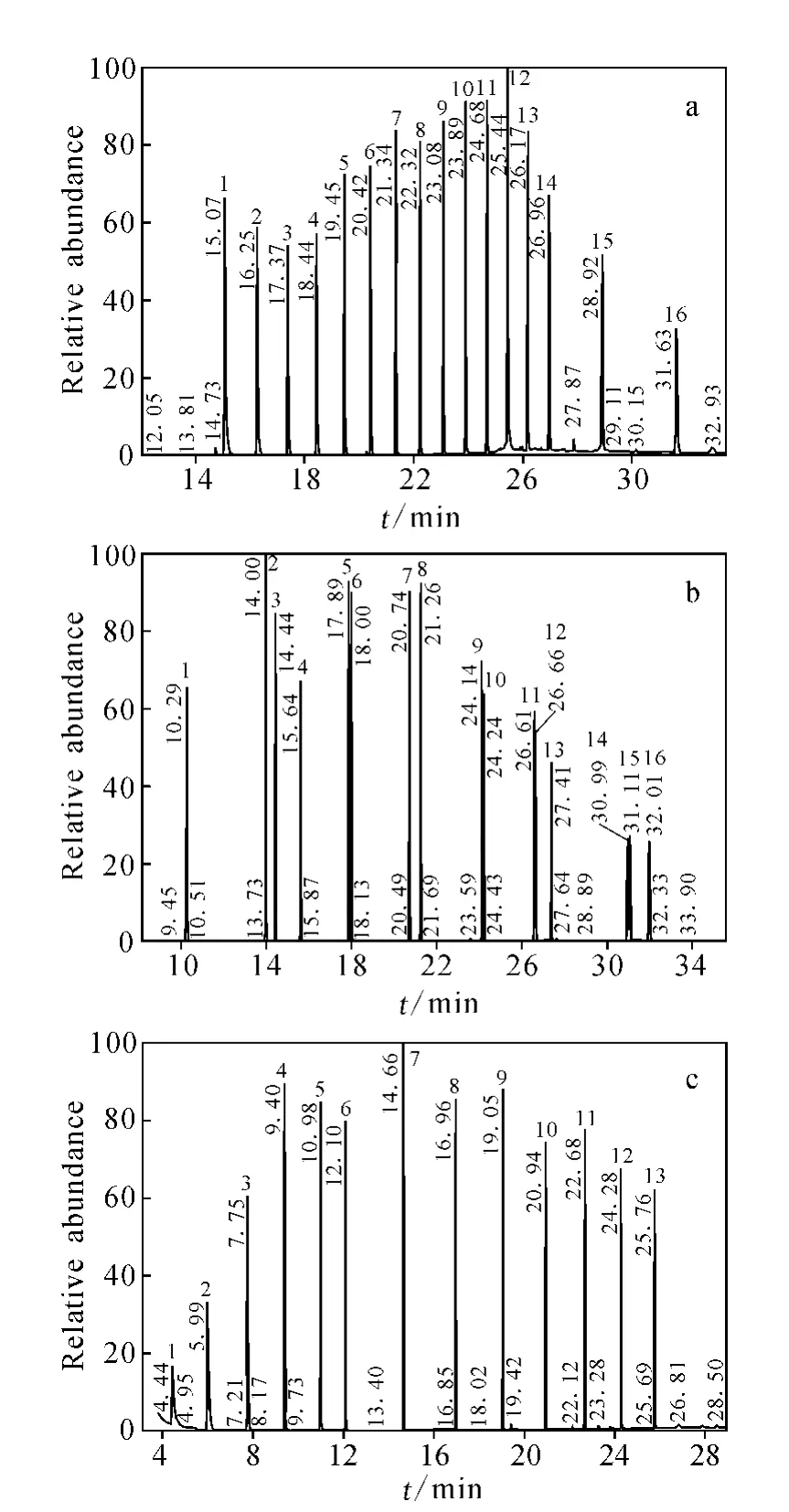
图1 正构烷烃(a)、多环芳烃(b)和有机酸酯(c)标准样品的色谱图Fig.1 The chromatogram of standard samples of alkane(a),PAHs(b)and organic esters(c)
2 实验结果与讨论
2.1 萃取条件的优化
大气颗粒物中有机组分含量一般较低,而且由于组成复杂,在分析过程中,不同种类之间还可能产生干扰,因此,利用色谱进行测定之前需要对目标组分进行分离富集,建立合适的萃取方法是基础性的一步。常用的颗粒物样品提取方法包括真空升华法,索氏提取法,超声萃取法和加速溶剂萃取法。其中,超声萃取法操作简单且仪器易得,萃取效率较高,目前应用较多。根据现有的实验室条件,本研究选择超声萃取作为颗粒物样品中有机组分的提取方法。
萃取溶剂的选择应根据相似相溶原理,要求萃取溶剂与目标物质的极性相似,从而提高萃取效率,并利于随后的分离净化。本研究中的三类有机组分正构烷烃、多环芳烃和有机酸分别呈非极性,弱极性和中强/强极性,在溶剂选择时应该综合考虑。实验室常用的有机溶剂包括正己烷、二氯甲烷、甲醇、丙酮、苯和甲苯等,Zheng等[3]的研究采用二氯甲烷作为萃取溶剂。根据文献报道以及综合考虑极性、毒性和后续处理中旋转蒸发的难易等因素,本研究选用正己烷和二氯甲烷作为萃取溶剂。定量移取3种组分的标准物质以及代用标准滴加到空白石英滤膜上,待稍微自然晾干后,依次用正己烷和二氯甲烷超声萃取,以确定溶剂用量和萃取次数。由于超声振荡时间过长会使振荡器内水温升高而导致某些组分损失[7],因此本实验的振荡时间为 30~35 min。实验平行5次,取平均结果。
实验表明,16种正构烷烃以及16种PAHs中的大多数在20 mL正己烷中已经基本被提取出来(80%),少量的可以进一步被 CH2Cl2提取。有机酸由于极性较强,在超声振荡作用下被正己烷提取的大约仅占15%,而绝大多数可以溶解在 CH2Cl2中,二元酸则需要用 20 mL CH2Cl2至少提取3次。为了使3种有机组分都能得到较高的萃取效率(>90%),本实验的颗粒物样品提取条件如下:用20 mL正己烷萃取2次,然后继续用20 mL CH2Cl2萃取3次,5次萃取液过滤后合并。合并后的萃取液经旋转蒸发至约3 mL,然后用N2吹扫近干,加入14%BF3/ CH3OH进行衍生。
2.2 衍生条件的选择与优化
由于强极性、难挥发的特点,有机酸通常难以直接用 GC或 GC/MS测定。有机酸的酯化反应克服了这一难题,通过反应将有机酸中的羧基转化成酯基,使分子中的氢键被破坏,从而极性降低、挥发性增大。三卤化硼催化下的有机酸与醇类的酯化反应容易进行,速率快、操作简单,适用于空间位阻大的羧酸的酯化。但应该控制该反应的条件不能太剧烈,否则可能会有少量的副反应。
综合考虑衍生化反应的各因素,即反应条件、反应速率以及衍生化试剂的易得和成本等,本实验选用14%BF3/CH3OH作为衍生化试剂,使之先与有机酸标准溶液反应,探索不同的反应条件对衍生化效率的影响,然后将反应条件优化,使之应用于气溶胶样品。在BF3的催化下,有机酸在室温下即可与甲醇进行酯化反应,但所需的反应时间较长;若提高反应温度,尽管反应时间缩短,但有可能带来副反应[8]。关于衍生化反应的温度条件,相关报道不尽相同, Zheng等[3]选择在85℃进行有机酸衍生化。
本实验比较了室温(25℃)、40℃和80℃3个温度条件下有机酸的衍生化效率,以确定最佳反应温度。同时,为了得到尽可能高的衍生化效率,在确定反应温度后,应该选取最佳的反应时间。反应时间太短反应不完全,衍生化效率低;时间过长则可能造成衍生产物的挥发或者水解。取一定量有机酸标准溶液与14%BF3/CH3OH分别反应5 min、20 min、45 min、60 min、180 min、24 h,比较衍生化效率,以确定最佳反应时间,实验平行4次。
由图2可以看出,在40℃时,多数有机酸转化率已经达到最大值,继续升高温度并不能提高转化率。考虑到本研究采用先衍生、后分离的流程,如果衍生化温度太高,溶液中的PAHs等可能会挥发而影响回收率,因此,采用40℃为最佳衍生化温度。通过不同反应时间下有机酸酯转化率的比较,发现在45 min时转化率已经基本稳定,而且在比较长的时间内,有机酸酯未发生分解,因此,采用45 min为最佳衍生化时间。

图2 有机酸衍生温度和衍生时间的选择Fig.2 The comparison of different derivatimation condition(time and temperature)of organic acids
2.3 净化、分离条件的选择与优化
衍生完毕,样品浓缩后需要将正构烷烃、PAHs和有机酸酯三类有机组分进行净化与分离,这是有机组分分析方法中的重要一环。由于大气颗粒物样品基体复杂,待测组分浓度低,经过超声萃取下来的部分中除了目标化合物,不可避免地带有其他杂质成分。这些杂质在用 GC或GC/MS分析时,可能会对目标化合物的出峰情况产生干扰或使检出率下降。此外,过多的杂质会降低色谱柱分离效果以及检测器灵敏度,甚至损坏仪器或缩短仪器的使用寿命。因此,要得到准确的、重现性好的数据,建立合适的净化分离条件是至关重要的。
固相萃取(solid phase extraction,SPE)是近些年发展起来的一种样品预处理技术,主要用于水体或土壤中痕量组分的富集与净化,在气溶胶领域的应用报道较少[9-11]。SPE法可以同时处理几个到几十个样品,利用固相萃取真空装置,流量控制易于实现。选择合适的溶剂将吸附在SPE柱上的样品萃取液洗脱为不同级分达到分离目的,通过条件优化可以实现较高的回收率。
2.3.1 固相萃取柱及其条件的选择优化 影响固相萃取方法的因素很多,如固定相的选择、柱子活化、洗脱速度等都会对目标组分回收率产生影响。固定相的选择依据2个因素:目标组分的极性和萃取样品所用溶剂的极性。应该尽量选择与目标组分极性相似的固定相,同时,固定相应该比所用萃取溶剂极性强,若溶剂极性太强,分析物在固定相上将得不到很好的保留。固定相分为正相与反相,蒋敏等[12]曾用反相固定相C18分离净化沉积物中的多环芳烃,Limbeck等[11]则用C18富集分离气溶胶中的有机酸。多数研究中仅用SPE研究某一类化合物,考虑的因素比较少,本实验希望选择一种固定相来实现三类有机污染物的分离与净化。由于本实验中萃取溶剂为非极性或弱极性,因此考虑采用正相固定相硅胶。
本实验先后比较了Supplco生产的500 mg与2 g两种型号SPE-Si柱的分离情况,发现这两种容量的柱子在上样后,用正己烷洗脱正构烷烃的同时,有大量多环芳烃被洗脱下来。这可能是在大量溶剂洗脱正构烷烃时,由于硅胶量太少,多环芳烃与吸附剂之间的吸附/解吸平衡被破坏,而随洗脱剂流出。反之,若柱床内硅胶足够多,可以重新建立吸附/解吸平衡。本实验采用自填SPE-Si柱,填柱方法是取10 g活化后的硅胶沿内壁倒入底部垫有玻璃棉的玻璃管内(直径15 mm),敲打外壁使之充实,然后在柱顶部加约1 cm无水Na2SO4,最后用约30 mL正己烷活化Si柱。实验表明,用上述自填SPE-Si柱能够实现正构烷烃、多环芳烃和有机酸酯的完全分离。
2.3.2 洗脱溶剂及用量的选择 洗脱流速是影响目标组分回收率的重要因素之一。蒋敏等[12]已经做了大量工作探讨洗脱流速对回收率的影响,过量的流速会导致回收率降低,一般应尽可能使用小的流速以保证高的回收率,但流速过小会造成洗脱时间过长。根据相关文献报道[12],本实验选择洗脱流速为2 mL·min-1。
合适的洗脱溶剂应该既能实现某一类目标组分的洗脱,又不至于将其他目标组分一并洗下来。根据相似相溶原理,同目标组分极性相近的洗脱剂洗脱效率最大。正构烷烃是非极性分子,而其他两类组分都具有一定极性,因此本研究采用正己烷作为正构烷烃的洗脱剂。实验表明,当用正己烷洗脱正构烷烃时,并没有破坏 PAHs在SPE硅胶柱上的保留,当用量为30 mL时,已经基本能把16种正构烷烃完全洗脱,回收率大于90%,示于图3a。
PA Hs具有弱极性,而且随环数增加,极性减弱,选用弱极性的CH2Cl2作为洗脱剂比较合适,但应考虑有机酸酯是否被同时洗脱下来。本研究以正己烷、CH2Cl2为基础,配制一定比例的洗脱剂,以期能完全洗脱PA Hs,同时又不影响有机酸酯在SPE柱上的保留。实验表明,当二者比例为20%时,约有1/2 PAHs的洗脱率已经达到80%,尚有1/2由于与硅胶颗粒之间的结合力比较大,而仅有部分被洗脱;当CH2Cl2比例为40%时,所有 PAHs的回收率都达到85%以上;继续增大比例,回收率变化不大。因此,本实验以CH2Cl2与正己烷配比为40∶100 (即40%CH2Cl2)作为PAHs洗脱剂。图3b表明,仅Pyr、Flu等几种3环、4环多环芳烃在40%CH2Cl2用量为10 mL时,峰值最大;而多数多环芳烃在用量为20 mL时达到洗脱峰值, 30 mL时已经基本被洗脱完毕。此过程中,有机酸酯基本未被洗脱,示于图3c。
Zheng等[3]采用V(乙醚)∶V(正己烷)= 1∶9的混合溶剂洗脱硅胶(400目)层析柱上吸附的有机酸酯类。本实验采用同样溶剂洗脱SPE硅胶柱上的有机酸酯,但回收率比较低,大部分酯类没有被洗脱下来,因此采用极性较强的有机溶剂丙酮,洗脱曲线示于图3c。与正构烷烃的洗脱类似,有机酸酯呈现出比较一致的洗脱规律,在20 mL丙酮洗脱时呈现峰值,30 mL时已经基本被洗脱完毕。

图3 固相萃取分离正构烷烃(a)、多环芳烃(b)和有机酸酯(c)的洗脱曲线Fig.3 The dilution curve of alkane(a),PAHs(b)and organic esters(c)seperated by SPE
2.4 质量控制与保证
根据文献报道的气溶胶中各类有机组分的大致含量,以标准储备液为基础,配制5种不同浓度系列的标准工作液,在选择优化的实验条件下,进行GC/MS分析,以峰面积响应值与组分浓度绘制标准曲线,各组分峰面积与浓度值呈良好线性关系,绝大多数组分相关系数大于0.999,满足后续定量分析要求。
为了监控超声萃取、衍生化以及分离净化等环节可能带来的污染,在分析气溶胶样品前进行了全程空白实验、溶剂空白实验以及加标回收率实验。分别以蒽-d10、硬脂酸-d35为 PAHs与有机酸的代用内标物,在实验开始时滴加到滤膜上。结果表明,全程空白实验以及溶剂空白实验中,没有目标组分出现。加标回收率实验表明,该方法中三类组分回收率都在70%~130%之间,满足美国EPA方法的要求。
2.5 气溶胶样品中有机组分的定性定量分析
在已经建立的包括萃取、衍生化以及分离净化在内的样品预处理方法以及仪器分析条件的基础上,对2003年9月~2004年7月期间的中流量PM2.5样品进行了测定,采用外标与内标结合的方式进行定量。由于实验中的正构烷烃和有机酸酯标准样品单个化合物的种类有限,部分没有标准样品的化合物通过全扫描确定其保留时间,对比标准谱库定性,以其相邻的化合物进行定量(假设其响应比为1:1)。例如,C14烃根据C15烃标样浓度进行计算,脂肪酸C17根据C16酸标样浓度进行计算等。定量检出的化合物包括:C10~C34之间的25种正构烷烃;16种美国EPA优控 PAHs;35种有机酸,包括 diC2~diC11之间的10种二元酸,C10~C32之间的23种饱和一元酸和C18∶1、C18∶2两种不饱和一元酸。
PM2.5样品中正构烷烃,多环芳烃和有机酸酯的典型色谱图示于图4。不同季节3种组分的碳数分布以及质量浓度存在很大差别,相关定性、定量分析结果的讨论见文献[13-14]。
3 结 论
本研究建立了基于固相萃取(SPE)技术的大气气溶胶中有机组分的样品前处理和色谱分析方法。确定了有机酸最佳衍生化条件,衍生温度和时间分别为40℃、45 min;确定了正构烷烃、PAHs和有机酸酯的最佳分离净化条件;应用固相萃取技术(SPE)可以同时处理数10个样品,比传统方法处理容量显著提高。
在上述建立的样品预处理和分析方法的基础上,定量测定了北京市PM2.5样品中的三大类有机组分,包括C10~C34之间的25种正构烷烃、16种美国 EPA优控 PA Hs以及35种有机酸(二元酸 diC2~diC11、脂肪酸 C10~C32和C18∶1、C18∶2不饱和酸)。
[1]牛红云,黄 宏,高士祥,等.大气气溶胶中有机成分研究进展[J].环境污染治理技术与设备, 2005,6(2):10-15.
[2]白志鹏,李伟芳.二次有机气溶胶的特征和形成机制[J].过程工程学报,2008,8(1):202-208.
[3]ZHENG M,FANG M,WANG F,et al.Characterization of the solvent extractable organic compounds in PM2.5aerosols in Hong Kong[J].Atmos Environ,2000,34(17):2 691-2 702.
[4]郭雪清,王英峰,施燕支,等.采暖期北京大气气溶胶中有机酸的测定[J].分析测试学报,2007,26 (增刊):181-186.
[5]HE K,YANG F,MA Y,et al.The characteristics of PM2.5in Beijing,China[J].Atmos Environ,2001,35(29):4 959-4 970.
[6]余学春,贺克斌,马永亮,等.北京市PM2.5水溶性有机物污染特征[J].中国环境科学,2004,24 (1):53-57.
[7]余学春,贺克斌,马永亮,等.气溶胶水溶性无机物及有机物的离子色谱测定[J].环境化学,2004, 23(2):218-222.
[8]詹益兴,陈贻文,周继红,等.衍生气相色谱及应用[M].长沙:湖南大学出版社,1988.
[9]LISKA I.Fifty years of solid-phase extraction in water analysis-historical development and overview[J].J Chromatogr A,2000,885(1/2):3-16.
[10]贾瑞宝,孙韶华,刘德珍.用固相萃取技术富集水中多环芳烃[J].色谱,1997,15(61):524-526.
[11]LIMBECK A,PUXBAUM H.A GC-MS method for the determination of polar organic compounds in atmospheric samples[J].Intern J Environ A-nal Chem,1999,73(4):329-343.
[12]蒋 敏,谢孟峡,谢 芳.沉积物中有机成分的分析方法研究[J].北京师范大学学报,2002,38 (3):370-376.
[13]段凤魁,贺克斌,刘咸德.北京PM2.5中有机酸的污染特征及来源研究[J].环境科学学报,2009, 29(6):1 139-1 145.
[14]段凤魁,贺克斌,马永亮.北京PM2.5中多环芳烃的污染特征及来源研究[J].环境科学学报, 2009,29(7):1 363-1 371.
Extraction,Separation and Purification of Three Kind of Organic Species in Atmospheric Particulate Matter and the Measurements by GC/MS
DUAN Feng-kui,HE Ke-bin
(State Key Joint L aboratory of Environment Simulation and Pollution Control,Department of Environmental Science and Environment,Tsinghua University,Beijing100084,China)
Organic aerosols were sampled by applying medium-flow PM2.5sampler during September 2003 to July 2004 in Beijing.This study built an optimum condition on the basis of solid phase extract(SPE)for the pretreatment of three kinds of compounds:n-alkanes, PA Hs and organic acids.The derivatization reagent for organic acids was 14%BF3/ CH3OH,and the optimal derivatization time and temperature were 40℃and 45 min,respectively.By using the self-filled SPE-Silica gel column,the above three kinds of compounds were seperated completely,and the recovery ranged from 70%—130%.The PM2.5samples were determined quantitativly by using GC/MS,including 25 alkanes,16 priority controlled PAHs by USEPA as well as 35 organic acids.
atmospheric particulate matter;n-alkanes;PA Hs;organic acids;GC/MS;derivatization
O657.63
A
1004-2997(2010)03-0165-07
2009-12-16;
2010-03-29
高等学校全国优秀博士学位论文作者专项资金(No.2007B57),国家重点实验室专项基金(No.09Z04ESPCT)资助
段凤魁(1973~),女,山东潍坊人,博士,从事大气污染控制研究。E-mail:duanfk@mail.tsinghua.edu.cn
