柱前衍生化-HPLC检测丙戊酸钠血清浓度方法验证
2010-07-12曲秀梅雷力力张志国
曲秀梅,雷力力,方 芳,李 冬,张志国
(佳木斯大学附属第一医院,黑龙江 佳木斯 154003)
丙戊酸钠为一线广谱抗癫痫药物,因分子结构中没有能够产生紫外光谱吸收的结构,用一般的 HPLC法难于直接测定,文献报道多用毛细管气相色谱法、酶联免疫法、荧光偏振免疫法等。目前应用较多的是“柱前衍生化高效液相色谱法检测丙戊酸钠的血清含量”,为了开展丙戊酸钠临床血药浓度监测,对文献[1,2]报道的柱前衍生化-高效液相色谱检测方法进行了综合,经过改进的方法检测结果稳定、准确,可以投入临床使用。
1 仪器及试剂
P230Ⅱ高效液相色谱仪(大连伊利特分析仪器仪器有限公司);XW-80涡旋混合器(上海精科实业有限公司);岛津AUW220D十万分之一天平。
色谱纯甲醇:天津凯通化学试剂有限公司生产;丙戊酸钠对照品 (湖南省湘中制药有限公司提供,含量:99.8%,批号 090405)环己烷羧酸 (美国 Sigma公司 ,批号:08265PD,);ω-溴苯乙酮(国药集团化学试剂有限公司,批号:20090212);其它试剂均为分析纯。
2 方法及结果
2.1 色谱条件
色谱柱:Hypersil ODS2C18(4.6mm× 250mm,5μm);柱温:30℃;检测波长:248nm;流动相:甲醇:水 (75:25);流速:1.0mL˙min-1;进样量:15 μL。
2.2 试验溶液的制备
精密称取丙戊酸钠对照品37.6mg,置50mL容量瓶中,用 0.2mol˙L-1氨水稀释至刻度 (含丙戊酸钠 0.752mg˙ mL-1),摇匀,作为丙戊酸钠标准溶液。精密称取 50.2mg环己烷羧酸,置50mL容量瓶中 ,用 0.2mol˙L-1氨水溶解并稀释至刻度,再精密量取该液5mL,置 25mL容量瓶中,0.2mol˙L-1氨水稀释至刻度,摇匀,作为内标溶液(含环己烷羧酸 0.2008mg˙mL-1)。精密称取125.4mg ω-溴苯乙酮,置25mL容量瓶中,用乙腈溶解并稀释至刻度,摇匀 ,作为衍生化试液 (含 ω-溴苯乙酮 5.016mg˙mL-1),放置于冰箱中保存。
2.3 样品制备及衍生化
取 200μL血清置空白采血管中,加入 30μL内标溶液和20 μL丙戊酸钠标准溶液 ,再加入 200 μL 1mol˙L-1的硫酸溶液,振摇混匀,然后加入4mL正己烷振摇萃取3min,静置5min使充分分层,分离出上清液2mL于另一试管中,加入40μL衍生化试液和20μL三乙胺,漩涡混匀,于63℃水浴衍生 10min,然后在氮气流下蒸发至干,立即取出,用甲醇0.3mL漩涡溶解 ,作为血清样品溶液。
2.4 色谱行为
在本实验条件下,衍生试剂峰与丙戊酸钠及内标衍生物能很好地分离,血清中无明显干扰峰。内标衍生物保留时间为7.00min,丙戊酸钠衍生物保留时间为10.90min。色谱图见图1。

图1 HPLC色谱图
2.5 标准曲线的制作
按 2.3项下方法 ,分别制备 8份样品溶液 ,丙戊酸钠标准溶 液 加入 量 分别为:18.80、37.60、56.40、75.20、94.00、112.80、127.84、150.40 μg˙mL-1,按色谱条件分别进样 ,计算各自的标准浓度(Y)对其峰高与内标峰高比值(X)的线性回归,其线性关系式为 Y= 86.852X+ 1.9179,r= 0.9988,线 性 范 围 为 18.80μ g~ 150.40μg˙mL-1,定 量 下 限 为18.80 μg˙mL-1。
2.6 精密度及回收率试验
按 2.3项下方法,分别制备高中低三个浓度 (94.00、75.20、56.40 μg˙mL-1)血清样品溶液各 5份 ,连续测定 3d,以内标法计算其方法回收率及日内、日间精密度。结果见表1。
表1 精密度及回收率试验结果 ±s)
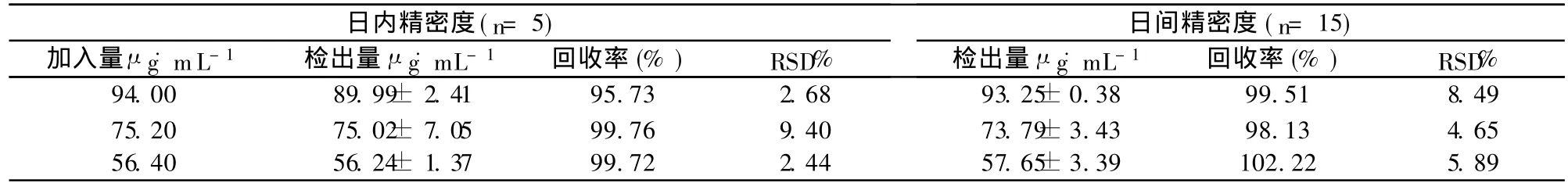
表1 精密度及回收率试验结果 ±s)
日内精密度(n=5)日间精密度(n=15)加入量μ g˙mL-1 检出量μ g˙mL-1 回收率(%)RSD%检出量μ g˙mL-1 回收率(%)RSD%94.00 89.99±2.41 95.73 2.68 93.25±0.38 99.51 8.49 75.20 75.02±7.05 99.76 9.40 73.79±3.43 98.13 4.65 56.40 56.24±1.37 99.72 2.44 57.65±3.39 102.22 5.89
2.7 稳定性试验
2.7.1 衍生物稳定性试验
按 2.3制备高、中、低三个浓度 (94.00、75.20、56.40 μg˙ mL-1)的样品溶液,分别于 0、24、48h时各进样 2次,以丙戊酸钠与内标峰面积比值计算 RSD,各样品 RSD为:0.82%;0.82%;1.30%。说明样品在48h内检测,结果稳定。
2.7.2 含药血清样品稳定性试验
取 血 清 9mL ,分 为 高、中、低 (94.00、75.20、56.40 μg˙mL-1)3个浓度组,各组分别加入丙戊酸钠标准溶液 375μL、300μL和 225μL,漩涡混匀 ,置冰箱冷藏 (2~ 10℃ )保存 ,然后于 0、5、10d分别取血清 200 μL各 5份 ,各加入 30μL内标溶液 ,从加入 200μL1mol˙L-1的硫酸溶液开始 ,按 2.3项下方法制备样品溶液。取水200 μL,从加入30μL内标溶液开始按2.3项下方法制备对照品溶液。按2.1色谱条件分别检测对照品溶液和样品溶液,记录色谱图。以内标法计算各样品回收率和各浓度回收率的 RSD。结果,高、中、低三个浓度样品回收率和 RSD分别为95.44±7.65,RSD=8.02%;93.92±3.37,RSD=3.59%;99.32±3.81,RSD=3.83%;说 明血清样品置冰箱(2~ 10℃)冷藏保存,10d内检测,结果稳定。
3 讨论
3.1 检测波长的确定
文献[1~4]报道检测波长有 266、248、235和 254nm,本文作者在以甲醇为溶剂,丙戊酸钠含量为0.752mg˙mL-1于200nm~280nm进行紫外扫描,上述各个波长处均有吸收,当浓度减少为0.2mg˙mL-1时,266nm波长处吸收很小 ,其他波长有吸收,随着波长减小,吸收度加大;当以同一浓度(0.752mg˙mL-1)样品 ,分别在 266、248、235和 254nm检测 ,丙戊酸钠及环己烷羧酸峰高按248、254、235和266nm依次递减,因而本文选择248nm作为吸收波长进行试验。
3.2 提取衍生溶媒的选择
文献报道提取衍生化溶媒有二氯甲烷[1]和正己烷[2],本文在用二氯甲烷作为溶媒,在248nm检测时,空白溶液在内标和丙戊酸钠保留时间处有吸收峰出现,改变流动相比例不能使杂质峰分开;在266nm处,空白、内标、丙戊酸钠和样品溶液均没有吸收峰出现,可能是检测浓度较低所致。因而选用正己烷作为衍生化溶媒。
3.3 蒸干终点的确定
在接近蒸干时,应注意随时观察,发现已蒸干立即取出,否则将会出现环己烷羧酸峰减小,丙戊酸钠峰加大现象。
3.4 计算方法的确定
内标法计算血清样品浓度,可以用峰高或峰面积[5]计算,试验结果显示以峰高计算样品浓度较以峰面积计算样品浓度准确,稳定,因而采用峰面积计算样品含量。
3.5 衍生化试剂用量的确定
有报道[6]衍生化试剂用量为2.5~ 10倍,本试验用量为7.33倍,当小于7.33倍时,线性关系试验中的高浓度部分会出现非理想峰高和峰形,因而选用本实验用量。
[1]孙志宏,阳国平,裴奇,等.2种丙戊酸钠片剂在人体内的相对生物利用度研究 [J].中南药学,2009,7(6):415
[2]陈坚,方维军,吴芳,等.高效液相色谱法测定丙戊酸血药浓度[J].中国临床药学杂志,2000,9(5):296
[3]展翔,徐枚.高效液相色谱法测定人血清中丙戊酸钠含量 [J].中国药业,2009,18(6):25
[4]石苏英,沈建幸.快速柱前衍生-高效液相色谱法测定丙戊酸血药浓度 [J].中国现代应用药学杂志,2007,24(4):315
[5]国家药典委员会编.中华人民共和国药典 [M].2005年版二部.北京:化学工业出版社,2005,28
