接骨康合剂的质量标准研究
2010-06-01夏德豪
夏德豪,胡 剑
(湖南省邵阳市药品检验所,湖南 邵阳 422001)
接骨康合剂为邵阳市中西医结合医院自制的医院制剂,处方由大黄、青皮、当归、红花、地龙等10味药材组成,具有行气、行血、化瘀止痛、接骨续筋的功效,用于治疗外伤淤血、扭伤、筋韧之伤、骨折初期等,经多年临床验证,疗效确切。为了有效地控制制剂质量,本试验采用薄层色谱(TLC)法对制剂中的大黄、青皮、当归进行定性鉴别,采用高效液相色谱(HPLC)法对大黄中的大黄素进行含量测定,报道如下。
1 仪器与试药
Waters高效液相色谱仪,包括1525泵、2996紫外检测器、1500柱温箱、717自动进样器;AG-135型电子天平(梅特勒-托利多〈上海〉有限公司)。大黄素对照品(批号为110756-200512,供含量测定用)、大黄对照药材(批号为120984-200602)、青皮对照药材(批号为121155-200502)、当归对照药材(批号为120927-200613)均购自中国药品生物制品检定所;接骨康合剂(邵阳市中西医结合医院自制);硅胶G(青岛海洋化工厂);甲醇为色谱纯,水为重蒸馏水,其他试剂均为分析纯。
2 方法与结果
2.1 TLC鉴别
大黄:取样品20 mL,用乙酸乙酯振摇提取3次,每次20 mL,合并乙酸乙酯液,将乙酸乙酯液蒸干,加水30 mL使溶解,用2%氢氧化钠溶液振摇提取3次,每次20 mL,合并氢氧化钠溶液,用浓盐酸酸化后,用乙酸乙酯振摇提取3次,每次20 mL,合并乙醚液,乙醚液蒸干,残渣加乙醇1 mL使溶解,作为供试品溶液。取去大黄的其他处方量药材,按制备工艺方法制成阴性样品,取20 mL同法制成阴性对照品溶液。另取大黄对照药材1 g,加甲醇10 mL,超声处理20 min,滤过,滤液蒸干,残渣加甲醇1 mL使溶解,作为对照药材溶液。照TLC法[2005年版《中国药典(一部)》附录ⅥB]试验,吸取上述3种溶液各10 μL,分别点于同一硅胶H薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15∶4∶1)的上层溶液为展开剂,展开,取出,晾干,置碘蒸气中薰约5 min,取出,待碘挥尽后,置可见光灯下检视。结果供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液无干扰(图1 A)。
青皮:取样品20 mL,用乙酸乙酯20 mL振摇提取,乙酸乙酯提取液蒸干,残渣加乙酸乙酯1 mL使溶解,作为供试品溶液。取去青皮的其他处方量药材,按制备工艺方法制成阴性样品,取20 mL同法制成阴性对照品溶液。另取青皮对照药材1 g,加水50 mL煎煮20 min,滤过,滤液用乙酸乙酯20 mL同上法操作,作为对照药材溶液。照TLC法[2005年版《中国药典(一部)》附录ⅥB]试验,吸取上述3种溶液各10 μL,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯(10∶6)为展开剂[1],展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视。结果供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照品溶液无干扰(图1 B)。
当归:取样品20 mL,用乙醚提取3次,每次20 mL,合并乙醚液,蒸干,残渣加乙酸乙酯2 mL使溶解,作为供试品溶液。取去当归的其他处方量药材,按制备工艺方法制成阴性样品,取20 mL同法制成阴性对照品溶液。另取当归对照药材1 g,加乙醚10 mL,超声处理10 min,滤过,滤液蒸干,残渣加乙酸乙酯1 mL使溶解,作为对照药材溶液。照TLC法[2005年版《中国药典(一部)》附录ⅥB]试验,吸取上述3种溶液各10 μL,分别点于同一硅胶G薄层板上,以环己烷-乙酸乙酯(4∶1)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照品溶液无干扰(图 1 C)。

2.2 含量测定
2.2.1 色谱条件
色谱柱:Kromasil C18柱 (250 mm ×4.6 mm,5 μm);流动相:甲醇 -0.2% 磷酸(75 ∶25)[2];检测波长:254 nm;柱温:35 ℃ ;流速:1.0 mL/min;进样量:10 μL。
2.2.2 溶液制备
精密称取大黄素对照品10.23 mg,置50 mL量瓶中,加甲醇适量使溶解并稀释至刻度,摇匀,精密量取20 mL,置100 mL量瓶中,加甲醇稀释至刻度,摇匀,作为对照品溶液(质量浓度为10.23 μg/mL)。精密吸取样品10 mL,置具塞锥形瓶中,精密加入三氯甲烷50 mL和10%盐酸溶液10 mL,密塞,称定质量,加热回流2 h,放冷,称定质量,用三氯甲烷补足减失的质量,摇匀,分取三氯甲烷液,精密量取20 mL,减压回收至干,残渣加甲醇使溶解,转移至10 mL量瓶中,并用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得供试品溶液。取除大黄外的其他处方量药材,按制备工艺方法制备阴性样品,按供试品溶液制备方法制成阴性对照品溶液。
2.2.3 方法学考察
系统适用性试验与阴性干扰试验:分别精密吸取对照品溶液、阴性对照品溶液与供试品溶液各10 μL,注入液相色谱仪,按拟订的色谱条件测定。结果大黄素与其他组分色谱峰可达基线分离,且与相邻色谱峰分离度大于1.5,理论塔板数按大黄素峰计均在3 000以上,表明色谱行为良好。供试品溶液色谱中,在与对照品溶液色谱峰相应的位置上检出色谱峰,而阴性对照品溶液在与对照品溶液色谱峰相应的位置无色谱峰,说明阴性样品对测定无干扰(见图 2)。
线性关系考察:分别精密吸取质量浓度为10.23 μg/mL的大黄素对照品溶液 1,3,6,12,20,25 μL,注入液相色谱仪,按拟订的色谱条件测定峰面积积分值,以对照品质量浓度为横坐标、峰面积积分值为纵坐标绘制标准曲线,得回归方程 A=2.43 × 106C+1 683.15,r=1.000 0(n=6)。结果表明大黄素进样量在0.010 23~0.204 6 μg范围内与峰面积积分值具有良好的线性关系。
精密度试验:取同一质量浓度的大黄素对照品溶液,在拟订的色谱条件下测定,连续进样5次。结果峰面积积分平均值为235 128,RSD=1.67%(n=5)。表明方法精密度良好。
稳定性试验:分别精密吸取同一供试品溶液10 μL,于0,2,4,8,16,24 h时进样。结果大黄素峰面积积分平均值为231 287,RSD=0.63%(n=6)。表明供试品溶液在24 h内稳定。
重现性试验:取同一样品(批号为080901)5份,按拟订的含量测定方法测定。结果大黄素平均含量为23.386 μg/mL,RSD=1.05%(n=5),表明方法重现性良好。
加样回收试验:分别精密量取对照品溶液(0.041 39 mg/mL)2.4,2.4,2.4,3.0,3.0,3.0,3.6,3.6,3.6 mL,置具塞锥形瓶中蒸干,各精密加入已知含量的供试品溶液5 mL(批号为080901,含量为23.386 μg/mL)和水5 mL,依法测定大黄素含量,计算回收率。结果见表1。
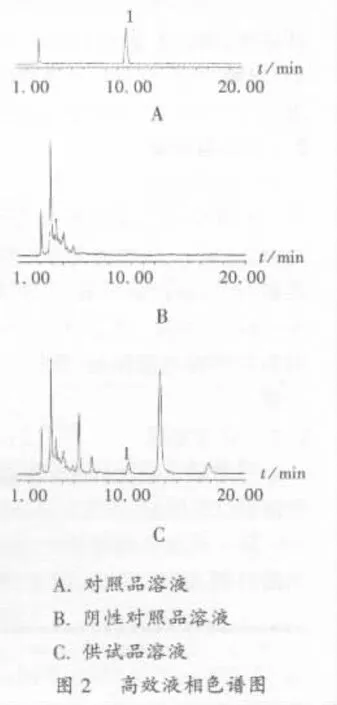

表1 大黄素加样回收试验结果(n=9)
2.2.4 样品含量测定
分别取对照品溶液和供试品溶液各10 μL进样,记录色谱图,按外标法计算大黄素含量。结果批号为080901,080902,080903的样品中大黄素平均含量为 23.39,31.56,29.53 μg/mL( n=3)。
3 讨论
方中大黄为君药,其主要有效成分为大黄素、大黄酸、大黄酚等蒽醌类化合物,而测定大黄素含量的HPLC法较成熟,故选择大黄中的大黄素作为含量测定对象。
方中干扰成分多,而青皮TLC鉴别的显色方法按文献[2]在紫外光灯365 nm下检视,只有1个斑点,且不清晰。试验中改用10%硫酸乙醇溶液显色,在105℃加热后,置紫外光灯(365 nm)下检视,专属性斑点增加至3个,且斑点清晰,分离效果好。
本方法薄层色谱和含量测定的阴性均无干扰,制剂质量基本稳定,为制剂质量控制提供了有效的分析方法,且方法简单,专属性强、重现性好,精密度、回收率都较满意,因此可作为接骨康合剂的质量控制方法。
[1]苗明三,李振国.现代实用中药质量控制技术[M].北京:人民卫生出版社,2000:603.
[2]国家药典委员会.中华人民共和国药典(一部)[M].北京:化学工业出版社,2005:17.
