紧密连接相关蛋白Claudin-5、ZO-1在大鼠蛛网膜下腔出血后早期脑损伤中的表达及意义
2010-05-25陈铎袁江伟宋磊魏翔泰关俊宏刘云会宗志红
陈铎,袁江伟,宋磊,魏翔泰,关俊宏,刘云会,宗志红
(中国医科大学 1.附属盛京医院神经外科,沈阳 110004;2.基础医学院生化教研室,沈阳 110001)
最新的研究结果显示,蛛网膜下腔出血(subarachnoid hemorrhage,SAH)后72 h内存在早期脑损伤(early brain injury,EBI),3~5 d 后发生脑血管痉挛(cerebral vasospasm,CVS),上述二者是造成SAH患者死亡或重残的最主要原因[1]。然而,EBI的确切分子病理机制尚不清楚,细胞凋亡机制可能参与了其发生、发展过程[2]。c-Jun 氨基端激酶(c-Jun N terminal kinase,JNK)信号转导通路参与了多种疾病的细胞凋亡过程[3]。脑损伤后发生的脑水肿病理改变与血脑屏障的开放密切相关,Claudin-5和ZO-1是最重要的脑血管内皮细胞紧密连接相关蛋白,它们的异常在血脑屏障开放中起关键作用[4,5]。本研究拟通过检测大鼠SAH后早期脑皮层微血管紧密连接相关蛋白Claudin-5和ZO-1的表达及JNK抑制剂对其表达的影响,进一步探讨SAH后早期脑损伤的机制。
1 材料与方法
1.1 实验动物及分组
健康成年雄性SD大鼠75只,体质量300~350 g,由中国医科大学附属盛京医院实验动物中心提供。采用随机数字表法将大鼠分为假手术组(sham组)、蛛网膜下腔出血(SAH)组、SAH+DMSO组、SAH+SP600125(10 mg/kg)组 和 SAH+SP600125(30 mg/kg)组,每组15只。SAH+二甲基亚砜(dimethyl sulfoxide,DMSO)、SAH+SP600125(10 mg/kg)和SAH+SP600125(30 mg/kg)组。在建立SAH模型基础上分别于实验前1h和实验后6h向腹腔内二次注入载体药物DMSO(Calbiochem公司)或JNK抑制剂SP600125(Sigma公司),每组大鼠中5只用于电镜形态学观察,10只用于Western blot检测Claudin-5、ZO-1的表达。
1.2 大鼠SAH模型的建立
采用血管内穿刺法建立大鼠蛛网膜下腔出血模型[6]:用 10%水合氯醛(300 mg/kg)经腹腔注射麻醉后,气管内插管,固定头部及四肢,做颈部正中3 cm切口,分离暴露右侧颈总动脉、颈内动脉、颈外动脉,电凝切断颈外动脉近分叉处分支(包括枕动脉、甲状腺上动脉及咽升动脉),分离结扎并切断颈外动脉,使颈外动脉近心端成一约5 mm长的残端,用2个无损伤动脉夹分别夹闭颈总动脉和颈内动脉,在颈外动脉起始段约3~5 mm处用眼科剪剪一“V”型小切口,下拉颈外动脉,与颈内动脉成一直线,再将1根长约5 cm的3-0尼龙线从颈外动脉上的“V”型小切口导入,经颈总动脉达颈内动脉,松开颈内动脉上的动脉夹,顺着颈内动脉向远端继续插入尼龙线至颈内动脉颅内段,有小的阻力感后再插入约2~3 mm刺破颈内动脉分叉部,迅速抽出尼龙线,用动脉夹夹住颈外动脉残端,松开颈总动脉上的动脉夹恢复血流,结扎颈外动脉残端,缝合颈部切口。术中股动脉插管接多导生理仪监测血压,维持肛温(37.5±0.5)℃,并定时测血气分析。sham组除了不刺破血管壁外,其余操作均与SAH组一致。
1.3 透射电镜观察大鼠脑皮层微血管超微结构
SAH模型建立24 h后处死大鼠,2%戊二醛溶液进行全身灌流,在冰盘内摘取大鼠脑皮质组织,迅速切成1 cm×1 cm×1 cm的小块,3%戊二醛溶液中预固定,0.1 mol/L磷酸缓冲液漂洗,1%四氧化锇固定,梯度乙醇脱水,定向包埋,定位后制成超薄切片,经醋酸双氧铀-枸橼酸铅双染色后,于透射电镜下观察、鉴别皮层神经元与皮层微血管组织,并照像。观察脑皮层微血管超微结构变化。
1.4 Western blot检测Claudin-5、ZO-1表达的变化
取各组大鼠新鲜大脑皮质,常规提取蛋白,10%SDS聚丙烯酰胺凝胶电泳分离蛋白样品,4℃转膜过夜,5%脱脂奶粉封闭;加入1∶500稀释山羊抗鼠多克隆抗体(Santa公司),4℃孵育过夜;二抗室温孵育2 h,DAB显色,照像。以GAPDH作为内对照。胶片扫描后,用Scion Image for Windows软件分析各条带的平均灰度值。
1.5 统计学分析
应用SPSS13.0统计软件。计数资料用率表示,采用t检验;计量资料用x±s表示,各组之间行ANOVA方差分析。P<0.05表示有统计学差异。
2 结果
2.1 大鼠SAH后24 h神经行为功能缺损评分
SAH后24 h对存活大鼠参照Bederson法[7]进行神经行为功能缺损评分,结果见表1。在SAH组和SAH+DMSO组中神经行为功能缺损评分集中在3 分和 4 分;SAH+SP600125(10 mg/kg)组集中在 2分和3分,而SAH+SP600125(30 mg/kg)组集中在1分和2分,说明JNK抑制剂SP600125可改善SAH存活大鼠神经行为功能缺损,SP600125(30 mg/kg)组尤为明显;sham组无死亡,SAH组、SAH+DMSO组、SAH+SP600125(10 mg/kg) 组、SAH+SP600125(30 mg/kg)组死亡率分别为30%、35%、25%、15%,各组间比较无统计学差异(P>0.05)。
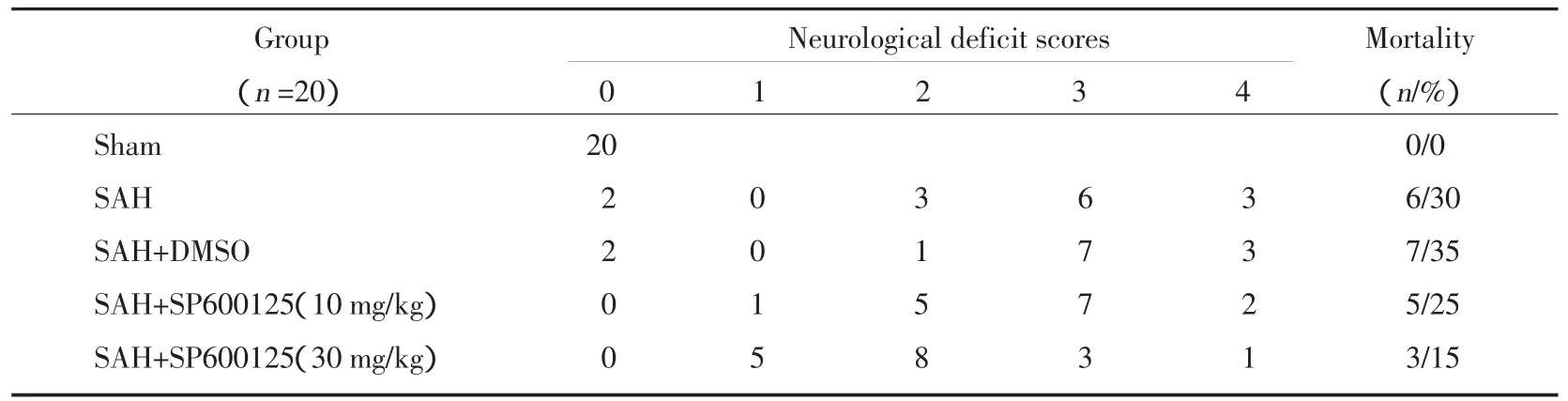
表1 SAH后24 h大鼠神经行为功能缺损评分及死亡率Tab.1 Neurological deficit scores and mortality after SAH
2.2 SAH大鼠脑皮层微血管组织形态学变化及SP600125抑制剂对其影响
透射电镜的结果(图1)显示,sham组大鼠脑皮层微血管内皮细胞呈梭形,胞膜连续完整,细胞核形状不规则,核膜尚完整,胞质内有较多核糖体、线粒体及粗面内质网等细胞器,相邻内皮细胞间可见紧密连接,血脑屏障未开放;SAH组和SAH+DMSO组血管内皮细胞多凸向管腔,细胞核形不规则,有切迹,核内异染色质明显边集,细胞基质密度降低,线粒体等细胞器减少,相邻内皮细胞间紧密连接明显松散,血脑屏障明显开放;SAH+SP600125(10 mg/kg)组细胞核形不规则,有切迹,核内异染色质明显边集,核膜部分模糊,细胞质内囊泡增多,相邻内皮细胞间紧密连接近腔面有部分松散,血脑屏障呈部分开放;SAH+SP600125(30 mg/kg)组细胞核形不规则,有切迹,细胞膜部分破坏,胞质内可见空泡,变性的线粒体、粗面内质网扩张,相邻内皮细胞间紧密连接呈部分松散状态,血脑屏障呈部分开放。

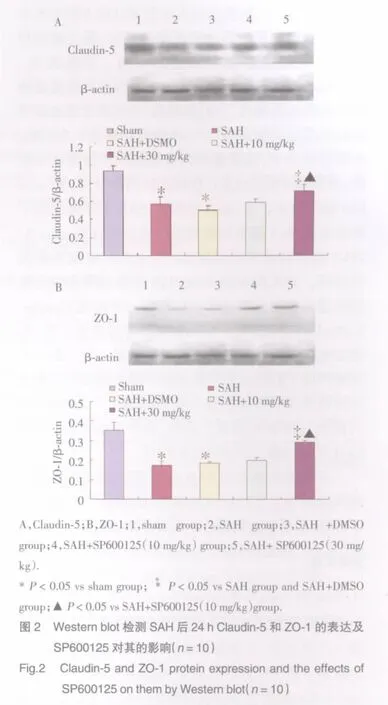
2.3 SAH大鼠脑皮层微血管紧密连接相关蛋白Claudin-5和ZO-1的表达及SP600125对其影响
Western blot结果(图2)表明,SAH组和 SAH+DMSO组Claudin-5和ZO-1表达水平下调,较sham组具有统计学差异(P<0.05);SAH组和 SAH+DMSO组之间比较,无统计学差异;与SAH、SAH+DMSO 组比较,SAH+SP600125(10 mg/kg)组Claudin-5和ZO-1的表达下调未受到明显抑制(P>0.05);SAH+SP600125(30 mg/kg)组 Claudin-5 和 ZO-1 的表达下调则受到明显抑制,10 mg/kg与30mg/kg SP600125两组间比较具有统计学差异(P<0.05)。
3 讨论
3.1 SAH后早期脑损伤
自发性蛛网膜下腔出血后早期脑损伤是指SAH后72 h内发生的一系列脑组织损害[8]。长期以来众多学者研究的重点多集中在SAH后3~5 d延迟发生的脑血管痉挛机制的研究,我们的前期研究结果也证实了迟发性脑血管痉挛是影响预后的关键因素之一[9,10]。然而,最近的研究结果提示,EBI也是造成患者死亡或重残的最主要原因之一[1]。然而,EBI发生、发展的机制目前尚不清楚,研究结果提示,细胞凋亡机制可能参与EBI的复杂分子病理形成过程。本研究结果证实,蛛网膜下腔出血后早期脑皮层血管内皮发生明显损伤改变,与细胞凋亡信号传导密切相关的JNK抑制剂SP600125可明显减轻血脑屏障形态破坏程度,并抑制脑皮层微血管紧密连接相关蛋白Claudin-5和ZO-1表达水平的降低,提示细胞凋亡机制可能参与了EBI复杂分子病理形成过程。
3.2 SAH后早期血脑屏障改变
研究显示,SAH后早期脑损伤病理生理表现为颅内压升高,脑灌注压和脑血流量下降,并引起急性缺血性改变,最主要病理表现为血管源性脑水肿,其机制尚不清楚。脑损伤继发血管源性脑水肿等改变的分子病理学基础是血脑屏障的破坏,最关键的核心环节是紧密连接相关蛋白结构、功能改变。Claudin-5、ZO-1是分布于脑微血管内皮间最重要的紧密连接相关蛋白,它们的结构、功能异常是血脑屏障破坏的最关键环节[4,5]。本研究结果进一步证实,在正常状态下,脑微血管内皮细胞间连接紧密无开放;蛛网膜下腔出血后,血脑屏障明显开放,Western blot结果显示,与sham组相比,SAH大鼠蛛网膜下腔出血后24 h脑皮层紧密连接相关蛋白Claudin-5、ZO-1的表达水平明显降低(P<0.05),血脑屏障受到破坏。JNK抑制剂SP600125可明显改善脑皮层微血管及神经元形态学损伤程度,并抑制Claudin-5、ZO-1的表达降低,即抑制血脑屏障开放程度,与透射电镜观察到的超微结构的变化相一致。提示大鼠蛛网膜下腔出血后早期血脑屏障结构蛋白发生破坏性改变是早期脑损伤关键性机制之一;SP600125可能通过抑制细胞凋亡通路,保护血脑屏障,发挥SAH早期脑损伤的神经保护作用。
本研究结果表明SAH后早期存在以血脑屏障结构破坏为主要特征的脑损伤,细胞凋亡可能参与了SAH后早期脑损伤分子病理学形成过程。
[1]Tseng MY,Czosnyka M,Richards H,et al.Effects of acute treatment with pravastatin on cerebral vasospasm,autoregulation,and delayed ischemic deficitsafter aneurysmal subarachnoid hemorrhage:a phase IIrandomized placebo-controlled trial[J].Stroke,2005,36(8):1627-1632.
[2]Sehba FA,Bederson JB.Mechanismsof acutebrain injury after subarachnoid hemorrhage[J].Neurol Res,2006,28(4):381-398.
[3]RepiciM,Borsello T.JNK pathway as therapeutic target to prevent degenerationinthecentralnervoussystem [J].AdvExp Med Biol,2006,588(1):145-155.
[4]Wolburg H,Lippoldt A.Tight junctions of the blood-brain barrier:development,composition and regulation[J].Vascul Pharmacol,2002,38(6):323-337.
[5]Anderson JM.Molecular structure of tight junctions and their role in epithelial transport[J].News Physiol Sci,2001:16(6):126-130.
[6]Schwartz AY,Masago A,Sehba FA,et al.Experimental models of subarachnoid hemorrhage in the rat:A refinement of the endovascular filament model[J].JNeurosci Methods,2000,96(2):161-167.
[7]Bederson JB,Pitts LH,Tsuji M,et al.Rat middle cerebral artery occlusion:evaluation of the model and development of a neurologic examination[J].Stroke,1986,17(3):472-476.
[8]Kusaka G,Ishikawa M,Nanda A,et al.Signaling pathways for early braininjuryaftersubarachnoidhemorrhage[J].JCerebBloodFlowMetab,2004,24(8):916-925.
[9]Chen D,Nishizawa S,Yokota N,et al.High-dose methylprednisolone prevents vasospasm after subarachnoid hemorrage through inhibition of proteinkinase Cactivation[J].Neurological Res,2002,24(3):215-222.
[10]Chen D,Chen JJ,Yin Q,et al.Role of ERK1/2 and vascular cell proliferation in cerebral vasospasm after experimental subarachnoid hemorrhage[J].Acta Neurochir,2009,151(9),1127-1134.
