Single Molecule Fluorescence Resonance Energy Transfer and Ensemble Biophysical Characterization of a G-quadruplex Formed in the Promoter of Human Myocyte Enhancer Factor 2D
2010-03-06ZHOUWenHuaYINGLiMing
ZHOU Wen-Hua YING Li-Ming,2,*
(1Molecular Medicine,National Heart and Lung Institute,Imperial College London,London SW7 2AZ,United Kingdom; 2Chemical Biology Centre,Imperial College London,London SW7 2AZ,United Kingdom)
Since the discovery of the iconic DNA double helical structure(B-form DNA)by Watson and Crick more than 50 years ago[1],other non B-form DNA secondary structures within certain sequences have been identified,such as left-handed DNA(ZDNA),triplexes(H-DNA,and sticky DNA),cruciforms,slipped hairpins,G-quadruplexes(also called G-tetraplexes,or G4 DNA), and i-motifs(i-tetraplexes)[2-4].It is believed that the majority of genome DNA is in the B-form,however,other secondary structures as mentioned above may also transiently form in many regions of the genome.These dynamic forms of structures are determined by DNA sequences,topology,DNA binding proteins, and other modifications of DNA[5].Increasingly,evidence indicates that these transient secondary structures relate to certain biological functions.This is plausible,as around 98%of human genomic DNA are non-coding sequences that would have certain biological functions in the view of evolution.Since the discovery that G4 can form in the G-rich regions of telomeric oligonucleotides in late 1980s[6],there have been a growing number of studies focusing on the structures and possible biological functions of these fascinating DNA motifs.
G4s are multiple vertically stacked guanine tetrads.Four guanines occupy each corner and form a G-tetrad plane,which is maintained by the Hoogsteen hydrogen bonds between N1,N7, O6,and N2 of each guanine base[7].As shown in Fig.1,G4s can be formed by four DNA strands(tetramolecular G4),two DNA strands(bimolecular G4),or single DNA strand(intramolecular G4).The side loop has three different types:lateral(L),external (E),and diagonal(D).2,3,or 4 planes stack together and form two,three or four-tetrad G4s.Monovalent cations,especially K+, can effectively bind to the core of G4s and further stabilize their structures,suggesting that the physiological conditions are more favorable for the formation of G4s[8].Although the core structure of G4 is very simple,the actual structures of G4 may vary and are difficult to predict.At the same time,the conformation of deoxyribose(C2′-endo orC3′-endo conformation)on the back bone and glycosidic bond angle also determine the overall structure of G4s(parallel or antiparallel).The structures of G4 are often heterogenous depending on the salt condition,for example,the same telomeric G4 motif can adopt several G4 conformations in dynamic equilibrium[9].

Fig.1 Schematic representation of G4 structures[8]
Although the majority of published work about the existence, structure,and biological relevance of G4 was carried out in vitro,more and more evidence has shown that G4s have special biological relevance and functions in living cells[10-13].The identification of G4 binding ligands and interacting proteins,such as TMPyP4(5,10,15,20-tetra(N-methyl-4-pyridyl)porphin),myogenic differentiation factor MyoD,the tetrahymena G4 binding protein,some telomeric proteins and the recently discovered G4R1(G4 resolvase)[14-15]suggest G4s provide a functional role in living cells.Furthermore,recent bioinformatics studies have demonstrate that the high density of putative G4-forming motifs in the transcriptional regulatory region(TRR-500 to+500)of genes across the human genome is associated with gene expression[16].Extensive studies carried out by Hurley′s group and others in the past decade have established the inhibitory roles of G4 sequences in promoter regions of many human genes,including the important proto-oncogenes c-myc and c-kit,HIF-1a,KRAS, Bcl-2,and growth factors PDGF-A and VEGF[17].There is therefore interest in G4s and their binding proteins are regarded as potential targets for anti-cancer drug design[18-19].G-rich motifs in mRNA were also found to be involved in regulation of gene translation[20],and specifically in tissue-specific alternative splicing by interacting with the hnRNP H protein subfamily[21-22].
In acute or chronic injury to the adult hearts,pathological responses,such as cardiomyocytes hypertrophy,and ventricular wall thinning,have been observed associated with development of pathological hypertrophy,which may result in heart failure and death[23-24].These pathological responses are actually caused by the activation of a complex signaling pathways that activate various transcription factors,including myocyte enhancer factor-2(MEF2),nuclear factor of activated T cells(NFAT),and GATA-4[25],leading to additional sarcomere assembly and expression of genes typical of the fetal cardiac gene program.There are four members(MEF2A-D)of the MEF2 family of MADS(MCM1, agamous,deficiency,serum response factor)box transcription factor family,which function as stress-dependent regulators of gene expression involved in pathological cardiac remodeling and multiple aspects of striated muscle development and disease.Recent studies about MEF2D in transgenic mice lacking the MEF2D gene found that the mice with a Mef2d loss-of-function mutation are viable,but display impaired response to stress signals that are normally associated with the development of cardiac hypertrophy,fibrosis,and fetal gene activation.However, the forced overexpression of MEF2D in mice can cause severe cardiomyopathy.These results suggest that Mef2d is possibly a key factor in regulating stress-dependent gene in adult heart, raising expectation that MEF2D might provide new target in developing“transcriptional”therapies for enhancing heart functions[26].Interestingly,in the TRR of MEF2D,we found four G4-forming motifs,which are at 232,169,85,and 18 bases upstream to the transcription start site(TSS;+1)of MEF2D from human(ID of transcript:NM005920).In particular,the motif starting at-169 contains four consecutive CCCC and its complementary strand would possibly form a stable 4-tetrad G4.In light of the previous studies about G4-forming motifs in the upstream TRR of c-myc,VEGF,HIF-1a,Ret,KRAS,Bcl-2,c-Kit, PDGF-A,and c-Myb[17],we suspect that this 4-tetrad G4 might play certain roles in regulating the expression of MEF2D in respect to the stress-dependent signaling,and therefore could be a target for the development of drugs to counter heart failure.
An understanding of G4 structures and dynamics is crucial to the further interrogation of their corresponding biological functions.The single molecule fluorescent resonance energy transfer (smFRET)technique has been proved to be a unique approach to resolving structure heterogeneity of DNA secondary structures such as G4[27-31].FRET is the non-radiative dipole-dipole energy transfer from a donor fluorophore to an acceptor fluorophore when they come within 1-10 nm of each other,and the FRET efficiency depends on the fluorophores′spectral overlap, separation and orientation[32].smFRET makes it possible to measure the conformational distribution of biomolecules under different conditions and to follow their folding/unfolding process in realtime[27].Herein we report our work using mainly smFRET as well as other ensemble biophysical methods to probe the existence,structures,and unfolding kinetics of a G4-forming motif found in the promoter of the human MEF2D gene.
1 Experimental
1.1 Materials
All labeled and unlabeled DNA oligos I-VI used in this study were purchased from IBA Biotechnology(Göttingen,Germany)and HPLC purified(double HPLC purification for labeled oligos).The sequences of these oligos are listed in Table 1.Oligos I and II,with Cy3 labeled at A47 and G60 respectively via a NHS ester,are the non-template strand fragment(-175 to-115, relative to the transcription start site of MEF2D)containing G4-forming sequence of human MEF2D promoter.Oligo III,with Cy5 labeled at T6 via a NHS ester,is complementary to the 30-nucleotide overhang of I and II.Oligo IV,with Cy5 labeled at T36 via a NHS ester,is complementary to I and II.Oligo V is complementary to the 30-nucleotide G4-forming region.VI is the unlabeled G4-forming sequence.The absorbance at 260 nm for DNA,550 nm for Cy3 and 650 nm for Cy5 was used to determine the concentration and the labeling ratio of all purchased DNA oligos.All labeled oligos have>95%purity.

Table 1 Details of oligos used in this study
1.2 Electrophoretic mobility shift assay
G4-forming and non-G4-forming DNA oligos were first dissolved in 10 mmol·L-1Tris-HCl buffer(pH 7.4)containing 100 mmol·L-1KCl to a strand concentration of 1 μmol·L-1.Prior to electrophoretic mobility shift assay(EMSA),the samples were heated to 95℃for 15 min and then slowly cooled down to room temperature over 4 h.Afterwards the oligos were loaded to a 12%native polyacrylamide gel(PAGE)in 1×Tris-borate-EDTA (TBE,pH 8.3)buffer for EMSA.Both the native PAGE and TBE buffer contained 100 mmol·L-1KCl.Control samples were heated to 95℃for 15 min,and then put on ice before being loaded onto the gel.Control experiments were carried out by loading the same samples onto 7 mol·L-1urea denaturing PAGE in 1×TBE buffer without KCl.All EMSAs were conducted at 120 V at room temperature.
1.3 CD characterization
Circular dichroism(CD)spectra were recorded on a J-810 CD spectrometer(JASCO,Essex,UK)at 20℃,using a 1 mm optical path length quartz cell and with a scan rate of 200 nm·min-1. All DNA samples were dissolved in 10 mmol·L-1Tris-HCl buffer(pH 7.4)containing 100 mmol·L-1KCl to a strand concentration of 12.5 μmol·L-1.The CD spectra were baseline corrected using blank buffer stated above.CD melting measurements were carried out with a temperature sweep rate of 1℃· min-1.Measured results were in milli-degrees and were converted into molar ellipticity((°)·cm2·dmol-1).
1.4 Single molecule FRET
A home-built dual-channel confocal fluorescence microscope was used to detect the conformational heterogeneity of G4 in buffer solutions[33-34].G4 samples were prepared by annealing of 100 nmol·L-1Cy3 labeled G4-forming DNA oligos with 150 nmol·L-1Cy5 labeled complementary strands in 10 mmol·L-1Tris-HCl buffer(pH 7.4)containing 100 mmol·L-1salts(LiCl, NaCl or KCl).Before single molecule measurements,samples were further diluted to 50 pmol·L-1in 10 mmol·L-1This-HCl buffer(pH 7.4)in the presence of 200 μmol·L-1Vitamin C,and 0.05%(φ,volume fraction)Tween 20 to reduce photobleaching and surface adsorption of DNA molecules.The donor,Cy3,was exited by a 150 μW collimated laser beam at 514 nm(argon ion, model 35LAP321-230,Melles Griot,Carlsbad,CA,USA).Fluorescent signals from donor and acceptor were collected by an oil-immersion objective(Apochromat 60×,NA 1.45,Nikon, Surrey,UK),detected by two single photon avalanche detectors (SPCM-ARQ-14,Perkin Elmer Optoelectronics,Salem,MA, USA)and recorded by two multichannel scalar cards(MCS-PCI, EG&G,ORTEC,Oak Ridge,TN,USA).FRET signals were identified from the background when the sum of photon bursts in the donor and acceptor channels exceeded a threshold of 40 photons per millisecond.Apparent FRET efficiency E of each FRET burst was calculated by the formula E=nA/(nA+γnD),where nA,nDare the acceptor and donor counts respectively,and γ is a constant used to compensate the differences that are due to quantum yields and optical detection efficiencies.γ was measured to be 1 for the Cy3/Cy5 pair.Cross-talk between Cy3 and Cy5 were determined to be 13%.FRET efficiency histograms were fitted by a 4-peak-Gaussian distribution model in Origin 7.0 (OriginLab Corporation,Northampton,MA,USA).Histograms obtained from six independent measurements were subject to global fitting to obtain the optimal sets of parameters for the four subpopulations observed.
1.5 G4 unfolding kinetics measurement
G4 unfolding kinetic of the end dual-labeled G4 system were measured at bulk level using the same instrument for smFRET as stated above.100 nmol·L-1G4 samples in 100 mmol·L-1K+solution(10 mmol·L-1Tris-HCl,pH7.4,200 μmol·L-1Vitamin C,and 0.05%(φ)Tween 20)were prepared as stated previously. Kinetics measurement were carried out by mixing Cy3 labeled G4 oligo II with its Cy5 labeled complementary strand oligo IV to a final concentration of 30 nmol·L-1for both.The mixing was carried out in a 4-well chambered coverglass(4 Chamber Borosilicate Coverglass System,Lab-Tek,Kamstrupvej,Denmark).The excitation laser power was reduced to 35 μW to avoid fluorescence signal saturation.Cy3/Cy5 signals were collected at 1 s interval for 12000 s.Solution background,cross talk between Cy3/Cy5 detection channels,and direct excitation of Cy5 were determined by blank buffer solution,Cy3 and Cy5 labeled samples,and were subtracted in the kinetics traces.All kinetics measurements were carried out at 25℃in a 1 mL volume of solution.
2 Results and discussion
2.1 Formation of G4 observed by EMSA
We first used a biochemical method,EMSA,to seek the evidence of G4 formation within oligo I in solution.For the native EMSA experiment,the mobility of DNA oligo is mainly determined by its size and charge.However,when two oligos(I and IV,both 60 bases in our experiment)have the same sizes,and therefore possessing similar charges,the relative mobility between them would be determined by their shapes(hydrodynamic diameter).The molecule with a more compact structure would migrate faster than the one with relaxed,loose structure.However,when ran on a denaturing gel in the presence of a DNA denaturing agent(urea,or less frequently,formamide),the mobility of denatured DNA is determined by its size,and almost completely independent of its base composition and sequence[35].As shown in Fig.2,on 12%native PAGE,the band corresponding to oligo I(lane 1)migrates much faster than the band from oligo IV(lane 2),while on the 12%denaturing PAGE containing 7 mol·L-1urea,these two bands have similar mobility(lanes 3 and 4 correspond to I and IV,respectively).These results indicate that oligo I can form a much more compact structure(s)than IV under native conditions(10 mmol·L-1Tris-HCl buffer containing 100 mmol·L-1KCl).We therefore attribute the slow migrating oligo I to the formation of G4.
2.2 Formation of G4 determined by CD spectroscopy
Since CD spectra have been well characterized for several G4 structures in solution,comparative CD analysis can provide primary evidence of the existence and structures of G4[36].Fig.3(a) shows that oligo VI incubated in 10 mmol·L-1Tris-HCl(pH 7.4)buffer containing either 100 mmol·L-1 KCl(upper curve) or LiCl(lower curve)both exhibit a CD spectrum characterized by a positive peak at 266 nm,a negative peak at 240 nm,and a pronounced positive shoulder between 280 nm and 310 nm.This spectral pattern is similar to that of the hybrid parallel/antiparallel G4 structure of Bcl-2 Pu39WT,whose structure has already been solved by NMR[37].Thus,these data suggest that the G rich region of VI could form hybrid parallel and antiparallel G4s,or form a mixture of pure parallel and antiparallel structures.In comparison to Pu48-mer,the 4-tetrad G4-forming sequence, critical to PDGF-A expression in the promoter of human plateletderivedgrowthfactorA(PDGF-A),theG4hereseemstobeeasier to form under less favorable conditions,suggesting it is more stable than its counterpart.As shown in Fig.3(a),the positive ellipticity at 266 nm in oligo VI in 100 mmol·L-1LiCl decreases only about 1.5 fold compared to that in 100 mmol·L-1KCl, while the same peak in Pu48-mer decreases more than 2.5 fold when changing from 100 mmol·L-1KCl to 100 mmol·L-1NaCl (Fig.2C in Ref.[36]).Secondly,the CD spectra of oligo VI in 100 mmol·L-1KCl and LiCl are exactly the same except for the different magnitude.On the contrary,a new antiparallel G-quadruplex structure forms in Pu48-mer in 100 mmol·L-1NaCl (Fig.2C in Ref.[36]).The exceptional stability of G4 structures of oligo VI was further verified by CD melting.As shown in Fig.3(b),the ellipticity value at 266 nm is almost independent of temperature until it reaches 70℃,and significant CD signals of hybrid parallel/antiparallel G4 structures of VI can still be detected even when the temperature was increased to 96℃.Due to the temperature limit of the CD instrument,we cannot determine the precise Tmof the oligo VI sample,but we estimate that it would be higher than 85℃.These results suggest that at least one G4 structure of VI should be more stable under thermal denaturing condition than its duplex form(Tm=74℃,estimated from nearest neighbor calculation method[38]).
2.3 Conformational analysis of G4 by smFRET
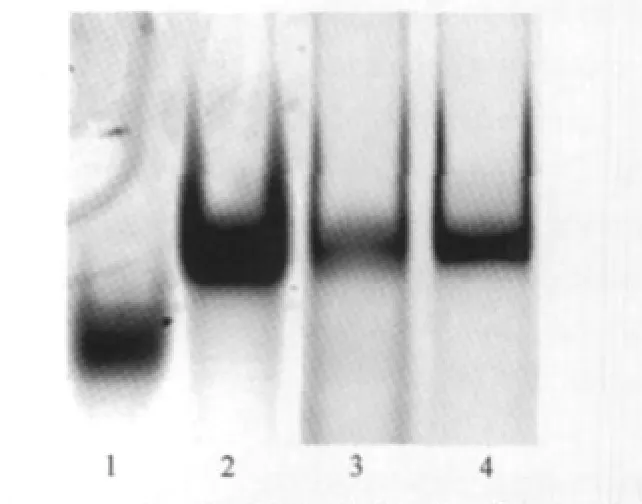
Fig.2 Native and denaturing gel electrophoresis indicating the formation of G4Lanes 1 and 2 are oligos I and IV run in a 12%native PAGE containing 100 mmol·L-1KCl.The mobility difference indicates the formation of G4 in oligo I.Lanes 3 and 4 are oligos I and IV run in a 12%denaturing PAGE containing 7 mol·L-1urea as controls.

Fig.3 CD spectra and CD melting curves of oligo VI(a) CD spectra of oligo VI in 100 mmol·L-1KCl(upper)and LiCl (lower);(b)CD melting curves of oligo VI in 100 mmol·L-1KClAll solutions contain 10 mmol·L-1Tris-HCl buffer(pH 7.4).ε:molar ellipticity

Fig.4 Schematic illustrating the designing principle of dual-labeled G4 systems for single molecule FRET(a)internal dual-labeled G4 system formed between oligos I and III;(b)end dual-labeled G4 system formed between oligos II and III;(c)duplex formed in the presence of oligo IV;and(d)duplex formed in the presence of oligos III and V. These systems were designed to show high FRET in folded states(a and b), and low or no FRET in duplex states(c and d).
The internal and end dual-labeled G4 systems composed of oligos I/III and II/III are shown in Figs.4(a)and 4(b)respectively.The G4 is connected to a 30 bp duplex on its 5′end.In both internal and end dual-labeled constructs,the acceptor,Cy5, is labeled to an adenine via a NHS ester 6 bp from the G4.In the internal dual-labeled system,the donor,Cy3,is labeled on a thymine in the third side loop of G4(Fig.4(a));and in the end labeled system,Cy3 is labeled on the last guanine involved in G4 (Fig.4(b)).In folded states of both internal and end dual-labeled systems,the donor-acceptor distances are within estimated Förster radius ofCy3/Cy5 pair,5.3 nm[39].In the presence of either V or IV,if G4 hybridizes into duplex state,it would lead to a large decrease of FRET efficiency from about 70%to less than 15%for the internal dual-labeled system,and negligible(<1%) for the end dual-labeled system,as a result of the increased separation between donor and accepter fluorophores in the duplex state(Figs.4(c)and 4(d)).
As the formation of G4 is dependent on metal ion coordination[40],initial smFRET studies were performed by using the Cy3/ Cy5 dual-labeled G4 systems to probe the formation of G4 in different salt solutions.Fig.5(a-d)shows the smFRET histograms of internal dual-labeled system (oligos I and III)obtained at 25℃in no salt and in 100 mmol·L-1Li+,Na+,and K+solutions,respectively.In these histograms,four subpopulations were identified by global fittings.S1 around zero is due to inactive acceptor[27]or unfolded DNA.S2,the broadest peak centered about 0.37,is likely the ensemble of partially folded quadruplex.S3 and S4,with their FRET efficiencies centered on 0.66 and 0.87 respectively,are assigned to two distinct G4 structures.One is likely parallel while the other antiparallel.However,we cannot rule out the co-existence of two hybrid parallel/antiparallel conformations.Low FRET S1 and S2 dominate in the histogram obtained in no salt solution(Fig.5(a)).In the absence of cations, the repulsion of central oxygen atoms would overwhelm the strength of Hoogsteen bond at N7 position of guanines involved in maintaining the overall stability of G4[41],while the coordination of cations between G-tetrad can reduce this repulsion,hence stabilize overall G4 structures.A general trend in alkali ions from the most stable to least is as follows:K+>Na+>Li+>compared to no salt.Thus,without central metal cations bound to the tetrads,unfolded and partially folded states would be dominant. In 100 mmol·L-1Li+solution,most of the unfolded states previously observed in the absence of salt(Fig.5(a),S1)were folded into high FRET G4s,which result in a smaller S1 and larger S3, S4(Fig.5(b)).Although the stabilizing effect of lithium cation is very weak,it can still promote G4 structures,evidenced by an increase of high FRET species in Fig.4(b).Histograms obtained in 100 mmol·L-1Na+and K+solutions(Figs.5(c)and 5(d))were identical,with fewer S2 and more S4.The larger increase of S4 over S3 is probably due to the different selectivity of the cation to the specific folded states[42].The similarity between the histograms obtained in Na+and K+solutions is possibly due to the spatial hindrance exerted by the bulky Cy5 label on the third side loop,preventing further structural changes caused by K+.

Fig.5 Single molecule FRET histograms in G4 conformational analysisHistograms obtained in smFRET conformational analysis experiments using internal(oligos I and III)and end(oligos II and III)dual-labeled G4 systems.(a)to(d),internal dual-labeled G4 system in the absence of salt,100 mmol·L-1Li+,Na+,and K+solutions respectively,measured at 25℃;(e)to(h),end dual-labeled G4 system in the absence of salt,100 mmol·L-1Li+,Na+,and K+solutions respectively,measured at 25℃.All solutions contain 10 mmol·L-1Tris-HCl(pH 7.4),200 μmol·L-1Vitamin C,and 0.05%(φ) Tween 20.
To learn the effect of labeling position on the folding and stability of G4,similar smFRET experiments were carried out on an end dual-labeled G4 system.The results are shown in Fig.5 (e-h).Global fitting did not work in this case as different G4s form under different salt conditions.Again,S1 is assigned to the inactive Cy5 or unfolded state,while S2 is an ensemble partially folded intermediates as described above.Without salt,only one high FRET subpopulation can be identified and S1 predominates(Fig.5(e)).In comparison to thatofinternal dual-labeled system in the absence of salt(Fig.5(a)),S2 population in the end dual-labeled system(Fig.5(e))is very small while a much larger and slightly shifted S1(“zero”peak)is observed,which is possibly caused by further separation between donor and acceptor in this system.The partially folded species may be merged into the“zero”peak and difficult to resolve by fitting alone.Histograms obtained from Li+and Na+solutions are almost identical(Figs.5(f) and 5(g)),indicating Li+and Na+may have similar effects on the structure and stability of these G4s.FRET histogram obtained in K+solution differs substantially from those obtained in either Na+or Li+,suggesting that K+favors the formation of a particular G4 conformation in S3 without the spatial hindrance exerted by the Cy5 label as observed in the internal labeled system.
2.4 G4/duplex competition assay using smFRET
To probe the stability of these G4 structures in the presence of their C-rich complementary strands,G4/duplex competition assays were carried out using smFRET.The Cy3 end-labeled G-rich strands(oligo II)were incubated with 1.5-fold of either III/V (3-component),or IV(2-component)Cy5 labeled C-rich complementary strands to allow the formation of full duplex.In the presence of C-rich strand complementary to the G4-forming sequence,an increase of S2,composed of intermediates is observed(comparing Fig.6(a),upper,to Fig.5(h)).This is likely due to the partial unfolding of G4 structures by the incomplete hybridization.In the 3-component system,a high FRET folded state S4 comes back and the other subpopulations remain relatively constant(comparing Fig.6(a),upper,to Fig.5(g)and 5H). In the 2-component system,this folded state S4 partially unfolds into intermediate S2,while the other folded state S3 still remains constant(comparing Fig.6(a)lower to upper).Thus,a G4/ duplex competition energy landscape of the end-labeled system can be proposed.As shown in Fig.6(b),on encounter of an adjacent C-rich complementary strand,the more stable G4 structure S3 first rearranges to a less stable G4 structure S4,which further unfolds into an intermediate before ending up as a full duplex.In this model,the rate-limiting step is the unfolding of G4 into duplex.
2.5 G4 hybridization and unfolding kinetics

Fig.6 Single molecule FRET histograms in G4/duplex competition assay and the proposed free energy landscape(a)Histograms obtained in smFRET G4/duplex competition assay of the end dual-labeled system.Upper:3-component(oligos II,III,and V)and lower:2-component(oligos II and IV).All experiments were carried out at 25℃in solutions containing 100 mmol·L-1K+,10 mmol·L-1Tris-HCl(pH 7.4),200 μmol·L-1Vitamin C,and 0.05%(φ) Tween 20;(b)Proposed free energy landscape.S3,S4 are two G4 structures,S2 are mainly intermediates between G4 and duplex and S1 is the full duplex.

Fig.7 A model for the competition between G4(oligo II)and duplex in the presence of full length C-rich complimentary strand(oligo IV)GQ is a G4 structure formed in the single stranded oligo II;C-Rich is the denatured single stranded oligo IV;GQC1 and GQC2 are schematic representations of two G4 structure corresponding to S3 and S4,respectively;P-Duplex is a partially formed duplex;and Duplex is the fully formed duplex.
In light of the energy landscape for the unfolding of the end dual-labeled G4 system in K+solution,we propose a kinetic scheme for the competition between G4 and duplex in the presence of C-rich complementary strands,as shown in Fig.7.In this model,GQ is the oligo II in G4 form,and C-Rich refers to the random-coiled complementary strand oligo IV.When oligos II and IV are incubated together,a partial duplex will quickly form in the 30-base long non-G4-forming region.These G4/duplex hybrids would initially maintain the most stable(lowest free energy)G4 structure GQC1,corresponding to the S3 species in Fig.5(h)and Fig.6(a).Following the progress of the reaction,some GQC1 would interconvert,via conformational rearrangement,into GQC2,a less stable G4 structure corresponding to the S4 species in Fig.5(h)and Fig.6(a).Afterwards,GQC2 unfolds,leaving a partially formed duplex(tagged as P-Duplex) that eventually anneals into a full duplex(tagged as Duplex).
Based on the facts that,i)GQC1 is more stable than GQC2;ii) the GQC2 population is much smaller than GQC1 in the presence of K+(Fig.5(h)and Fig.6(a),lower);and iii)the formation of the P-Duplex is very fast due to the existence of the adjacent complementary strand,the model proposed above can be simplified to,

where k1and k2are the partial annealing rate(duplex hybridization rate)and the G4 unfolding rate,respectively.From this reaction scheme,it is not difficult to obtain the rate equation against species GQC as follows:
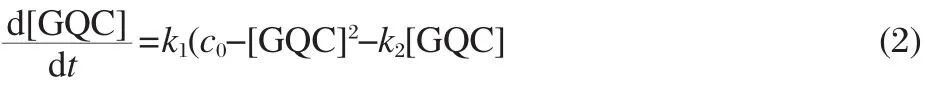
where c0is the initial concentration of both GQ and C.The analytical solution of Eq.(2)can be used to obtain the duplex hybridization rate and the subsequent G4 unfolding rate from fitting to the experimental data,as shown in Fig.8.The fitting was carried out in MatLab 2009(MathWorks,Inc.,Natick,MA,USA) by a built-in curve-fitting tool.The hybridization rate constant and unfolding rate constant were determined to be(3.8±0.2)×104mol-1·L·s-1and(8.3±0.4)×10-6s-1,respectively.In comparison to the hybridization rate constant of 1.37×105mol-1·L·s-1between a 31 base G4-forming strand and its complementary from the human c-myc promoter[43],the hybridization rate constant measured here is slower but well within the typical range of DNA hybridization.Remarkably,the unfolding rate constant of the G4 studied here is two to three magnitude slower than that of typical 3-tetrad G4s,with literature values 7.90×10-3s-1for human c-myc G4[43]and 2.34×10-2s-1for human telomeric G4[44], respectively.In addition to the results from single molecule duplex/G4 competition assay,the unfolding kinetics confirmed the exceptional stability of the four-tetrad G4,which could form easily in the promoter during gene transcription.

Fig.8 Kinetics of G4 unfoldingHybridization time trace recorded by acceptor Cy5 fluorescence(black)and the fit (red)based on the solution of Eq(2).The reaction was carried out at 25℃in 100 mmol·L-1KCl solution containing 10 mmol·L-1Tris-HCl(pH 7.4), 200 μmol·L-1Vitamin C,and 0.05%Tween 20.
3 Conclusions
A prerequisite for regulatory roles of G4 in gene expression lies in its capability of rivaling the duplex in the gene promoter. We observed the formation of exceptionally stable G4 structures in the GC-rich motif found in the promoter of human MEF2D. smFRET assay indicates these G4 structures can efficiently compete with the duplex,raising the expectation that this G4 is a molecular switch that regulates MEF2D expression,therefore it may serve as a new target for the development of drugs against MEF2D related heart malfunctions.Based on a kinetics scheme where G4 unfolding is rate-limiting,we obtained duplex hybridization rate and G4 unfolding rate,which confirms the sta-bility of the 4-tetrad G4.We also demonstrated that a fluorophore internally labeled in the third loop of this G4 could provide us a new option to monitor the formation and dynamics of G4 by FRET.This approach may be particular beneficial where there is a long loop within the G4 sequence[45]and the label would not interfere G4 formation.We are hoping to apply this labeling strategy to the study of G-rich minisatellites,where like telomeres,multiple G4s could form.It is worth noting that the sm-FRET research is focused on probing conformational dynamics and real-time kinetics of G4 structures at single molecule level, and the exact structures of these biomolecules formed can be determined by NMR spectroscopy,which is beyond the scope of this study.Nevertheless,DMS footprinting can provide valuable information about the involvement of base Gs in the quadruplex[11].
Acknowledgment: The authors wish to thank Dr.Nigel Brand for proofreading the manuscript.
1 Watson,J.D.;Crick,F.H.Nature,1953,171:737
2 Wells,R.D.J.Biol.Chem.,1988,263:1095
3 Wells,R.D.Trends.Biochem.Sci.,2007,32:271
4 Bacolla,A.;Wells,R.D.J.Biol.Chem.,2004,279:47411
5 Wells,R.D.;Blakesley,R.W.;Hardies,S.C.;Horn,G.T.;Larson, J.E.;Selsing,E.;Burd,J.F.;Chan,H.W.;Dodgson,J.B.;Jensen, K.F.;Nes,I.F.;Wartell,R.M.CRC Crit.Rev.Biochem.,1977,4: 305
6 Henderson,E.;Hardin,C.C.;Walk,S.K.;Tinocov Jr.,I.; Blackburn,E.H.Cell,1987,51:899
7 Simonsson,T.J.Biol.Chem.,2001,382:621
8 Bates,P.;Mergny,J.L.;Yang,D.EMBO Rep.,2007,8:1003
9 Li,J.;Correia,J.J.;Wang,L.;Trent,J.O.;Chaires,J.B.Nucleic Acids Res.,2005,33:4649
10 Ribeyre,C.;Lopes,J.;Boule,J.B.;Piazza,A.;Guedin,A.;Zakian, V.A.;Mergny,J.L.;Nicolas,A.PLoS Genet.,2009,5:e1000475
11 Siddiqui-Jain,A.;Grand,C.L.;Bearss,D.J.;Hurley,L.H.Proc. Natl.Acad.Sci.U.S.A.,2002,99:11593
12 Paeschke,K.;Simonsson,T.;Postberg,J.;Rhodes,D.;Lipps,H.J. Nat.Struct.Mol.Biol.,2005,12:847
13 Lipps,H.J.;Rhodes,D.Trends Cell Biol.,2009,19:414
14 Fry,M.Front.Biosci.,2007,12:4336
15 Creacy,S.D.;Routh,E.D.;Iwamoto,F.;Nagamine,Y.;Akman,S. A.;Vaughn,J.P.J.Biol.Chem.,2008,283:34626
16 Du,Z.;Zhao,Y.;Li,N.Genome Res.,2008,18:233
17 Qin,Y.;Hurley,L.H.Biochimie,2008,90:1149
18 Han,H.Y.;Hurley,L.H.Trends Pharmacol.Sci.,2000,21:136
19 Gryaznov,S.M.;Jackson,S.;Dikmen,G.;Harley,C.;Herbert,B. S.;Wright,W.E.;Shay,J.W.Nucleosides Nucleotides&Nucleic Acids,2007,26:1577
20 Arora,A.;Dutkiewicz,M.;Scaria,V.;Hariharan,M.;Maiti,S.; Kurreck,J.RNA,2008,14:1290
21 Arhin,G.K.;Boots,M.;Bagga,P.S.;Milcarek,C.;Wilusz,J. Nucleic Acids Res.,2002,30:1842
22 Bruce,S.R.;Dingle,R.W.C.;Peterson,M.L.RNA,2003,9: 1264
23 Czubryt,M.P.;Olson,E.N.Recent Prog.Horm.Res.,2004,59: 105
24 Frey,N.;Katus,H.A.;Olson,E.N.;Hill,J.A.Circulation,2004, 109:1580
25 McKinsey,T.A.;Olson,E.N.J.Clin.Invest.,2005,115:538
26 Kim,Y.;Phan,D.;van Rooij,E.;Wang,D.Z.;McAnally,J.;Qi, X.;Richardson,J.A.;Hill,J.A.;Bassel-Duby,R.;Olson,E.N. J.Clin.Invest.,2008,118:124
27 Ying,L.M.;Green,J.J.;Li,H.;Klenerman,D.;Balasubramanian, S.Proc.Natl.Acad.Sci.U.S.A.,2003,100:14629
28 Shirude,P.S.;Okumus,B.;Ying,L.M.;Ha,T.;Balasubramanian, S.J.Am.Chem.Soc.,2007,129:7484
29 Shirude,P.S.;Ying,L.M.;Balasubramanian,S.Chem.Commun., 2008:2007
30 Shirude,P.S.;Balasubramanian,S.Biochimie,2008,90:1197
31 White,S.S.;Balasubramanian,S.;Klenerman,D.;Ying,L.M. Angew.Chem.Int.Edit.,2006,45:7540
32 Selvin,P.R.Nat Struct.Biol.,2000,7:730
33 Ying,L.M.;Wallace,M.I.;Balasubramanian,S.;Klenerman,D. J.Phys.Chem.B,2000,104:5171
34 Wallace,M.I.;Ying,L.M.;Balasubramanian,S.;Klenerman,D. Proc.Natl.Acad.Sci.U.S.A.,2001,98:5584
35 Bartlett,J.M.S.;Stirling,D.PCR protocols.2nd ed.Totowa: Humana Press,2003:68
36 Qin,Y.;Rezler,E.M.;Gokhale,V.;Sun,D.;Hurley,L.H.Nucleic Acids Res.,2007,35:7698
37 Dai,J.;Dexheimer,T.S.;Chen,D.;Carver,M.;Ambrus,A.;Jones, R.A.;Yang,D.J.Am.Chem.Soc.,2006,128:1096
38 SantaLucia Jr.,J.Proc.Natl.Acad.Sci.U.S.A.,1998,95:1460
39 Bowen,M.E.;Weninger,K.;Ernst,J.;Chu,S.;Brunger,A.T. Biophys.J.,2005,89:690
40 Arnott,S.;Chandrasekaran,R.;Marttila,C.M.Biochem.J.,1974, 141:537
41 Mergny,J.L.;De Cian,A.;Ghelab,A.;Sacca,B.;Lacroix,L. Nucleic Acids Res.,2005,33:81
42 Sen,D.;Gilbert,W.Nature,1990,344:410
43 Halder,K.;Chowdhury,S.Nucleic Acids Res.,2005,33:4466
44 Green,J.J.;Ladame,S.;Ying,L.M.;Klenerman,D.; Balasubramanian,S.J.Am.Chem.Soc.,2006,128:9809
45 Cogoi,S.;Paramasivam,M.;Spolaore,B.;Xodo,L.E.Nucleic Acids Res.,2008,36:3765
