利用RAP-PCR检测高盐刺激下Shewanella piezotolerans WP3的基因差异表达
2009-12-02李升康汕头大学理学院生物系广东汕头515063国家海洋局生物遗传资源重点实验室福建厦门361005
李升康 (汕头大学理学院生物系,广东 汕头 515063;国家海洋局生物遗传资源重点实验室,福建 厦门 361005)
李金媛,王玉桥 (国家海洋局生物遗传资源重点实验室,福建 厦门 361005)
利用RAP-PCR检测高盐刺激下ShewanellapiezotoleransWP3的基因差异表达
李升康 (汕头大学理学院生物系,广东 汕头 515063;国家海洋局生物遗传资源重点实验室,福建 厦门 361005)
李金媛,王玉桥 (国家海洋局生物遗传资源重点实验室,福建 厦门 361005)
选取高盐刺激为条件,利用实验室模拟极端环境培养条件下的菌体提取RNA,采用差异显示方法RAP-PCR扫描盐刺激下基因的差异表达。结果表明,在高盐刺激下,WP3下调表达的基因是SS1: hypothetical protein; SS2: ferroxidase。上调的基因是SS3: gamme-butyrobetaine antiporter; SS4: hypothetical protein。
ShewanellapiezotoleransWP3;差异显示;高盐刺激;基因表达
深海环境是一个高压、低温、寡营养、高盐度的极端环境,生存在这样的极端环境下的生物体有着怎样的新陈代谢方式,一直是生物学科研工作者关注的焦点。深海极端微生物的研究主要集中在极端嗜压菌、极端嗜高温菌等方面,如PhotobacteriumprofundumSS9[1]、Aquifexaeolicus[2]。 差异显示方法是研究基因差异表达的常用方法。由于原核生物的mRNA缺少polyA尾巴,故微生物中研究基因差异表达的方法是RNA arbitrarily primed PCR (RAP-PCR)。鉴于RAP-PCR方法相对成熟,并且在微生物差异基因表达调控的研究中得到了广泛的应用[3,4],本研究运用RAP-PCR方法来扫描分离自深海的耐嗜压菌株ShewanellapiezotoleransWP3在高盐度刺激下的差异表达基因。
1 材料与方法
1.1 细菌的实验室培养及高盐刺激

表1 差异显示RAP-PCR所用的引物Table 1 The primers of differential display RAP-PCR
ShewanellapiezotolranceWP3分离自西太平洋的1 914 m深的深海沉积物样品中[5]。深海细菌WP3通过2216E实验室培养,待长至对数生长中期后换成12% NaCl的2216E培养30 min,并用高盐刺激前的菌体作为阳性对照。
1.2 RNA的提取及纯化
总RNA 提取采用TRIZOL 试剂(MBI 公司) 并按说明操作。取1 μL 在分光光度计上检测RNA 质量及浓度,取2 μL 在甲醛变性琼脂糖胶上电泳检测是否降解。
1.3 反转录
反转录遵照逆转录试剂盒(Invitrogen公司)
说明操作。所用反转录引物及PCR引物见表1。用前将T-RNA 稀释至0.1 mg/mL,加入0.1 mg/mL的T-RNA 2 μL和2 μmol/L的 RT primer 2 μL; 吹吸混匀(勿漩涡),短暂离心,70 ℃ 5 min(采用带热盖的PCR仪);接着冰上骤冷,短暂离心; 再依次加入ddH2O 9.3 μL、AMV逆转录酶缓冲液 (5×) 4.0 μL、dNTP (250 μmol/L) 2.0 μL,RNasin (40 U/μL)0.5 μL,AMV逆转录酶 (200 U/μL) 0.2 μL 至总体积20 μL。逆转录程序为42 ℃ 90 min,70 ℃ 15 min;4 ℃ 暂存。
1.4 RAP-PCR
在0.2 mL的薄壁管中加入ddH2O 1.95 μL、PCR缓冲液1.0 μL、MgCl2(25 mmol/L) 1.5 μL、dNTP (250 μmol/L)2.0 μL、5’PCR primer (2 μmol/L) 1.75 μL、3’RT primer(与逆转录混合物中的一致,5 μ mol/L)0.7 μL、逆转录混合物1.0 μL、Taq酶 (5 U/μL) 0.1 μL 至总体积10 μL。PCR程序(采用带热盖的PCR仪)为:95 ℃ 2 min,4个循环(92 ℃ 15 s,50 ℃ 30 s,72 ℃ 2 min);30个循环:92 ℃ 15 s,55 ℃ 30 s,72 ℃ 2 min,72 ℃ 7 min,4 ℃暂存。所有的PCR皆做双份,以验证重复性。同时须做以无RT酶的逆转录混合物为模板的对照。
1.5 电泳检测
将检测后的PCR产物加入变性的上样缓冲液10 μL,混匀后在PCR仪中95 ℃变性3 min,然后在冰中迅速冷却变性成单链。用8%的聚丙烯酰胺胶(100 mL)加入10%的过硫酸铵800 μL、TEMED 50 μL及尿素7.5 mol/L制胶,应用CBS Scientific电泳系统1 200 W恒压电泳4 h至二甲苯碃到达玻璃板的底部。采用通常的银染方法染色检测。
1.6 差异片段的回收、重扩增及反向Northern杂交鉴定
用煮沸法从测序胶的泳道中将具有明显差异的条带依次回收并在相应的差异显示反应条件下进行重扩增。用1%的琼脂糖凝胶电泳检测,用琼脂糖纯化试剂盒纯化,纯化后一部分制备探针。准备好纯化的RNA,反转录合成cDNA ,PCR检验cDNA 质量合格后纯化,用地高辛标记(按试剂盒提供的方法检测标记效果) ,取回收的电泳差异带重扩增的PCR 产物50 ng,94 ℃变性5 min,点样器将样品点到规划好的尼龙膜上,进行紫外交联,按25 ng/mL取标记好的探针,在杂交炉杂交,随后进行免疫检测,检测所回收差异片段的真假。
1.7 差异片段克隆测序及比对
将经过Northern 杂交鉴定的阳性片段连接到载体pGEM-T Easy Vector (Promega 公司),并转化到大肠杆菌E.coliDH5α菌株,挑选白色菌落,经菌落PCR 验证的阳性克隆在含氨苄青霉素的LB 培养基培养。取1.5 mL菌液送交上海生工公司测序。取1.5 mL用质粒提取试剂盒(TaKaRa 公司)提取质粒,保存备用。测序结果在GenBank中进行同源性分析。
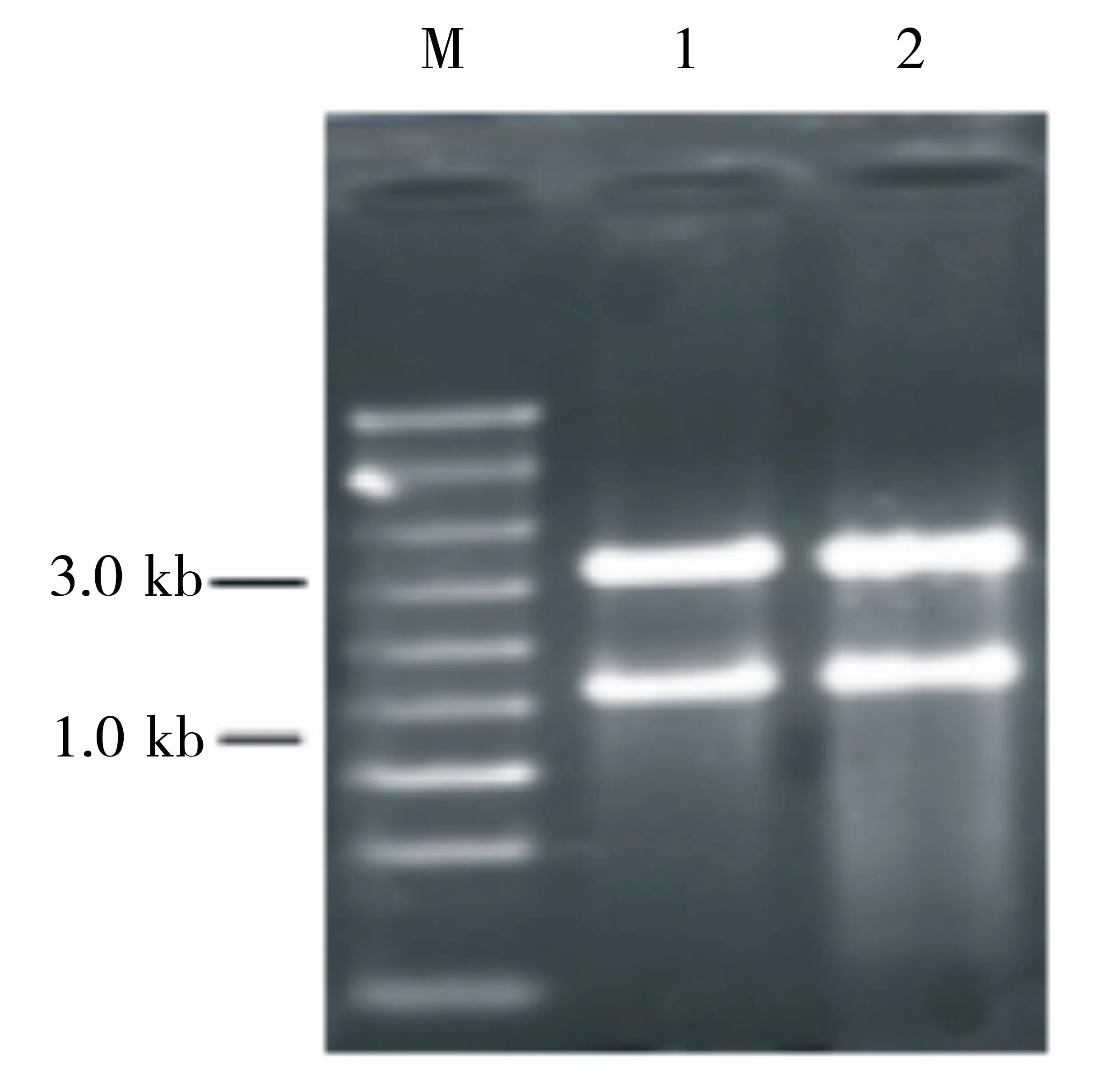
1. 阳性对照:正常生长的WP3总RNA;2. 高盐刺激下的WP3总RNA;M:RNA Marker图1 RNA提取电泳效果图Figure 1 Agarase gel electrophoresis of RNA extract
2 结果与分析
2.1 WP3菌体的总RNA的提取
RNA的电泳图片见图1。从图1可以很清晰地看到16S,23S 2条带,且23S的量约为16S的2倍。证明所提取的RNA基本无降解,可以进行进一步纯化。
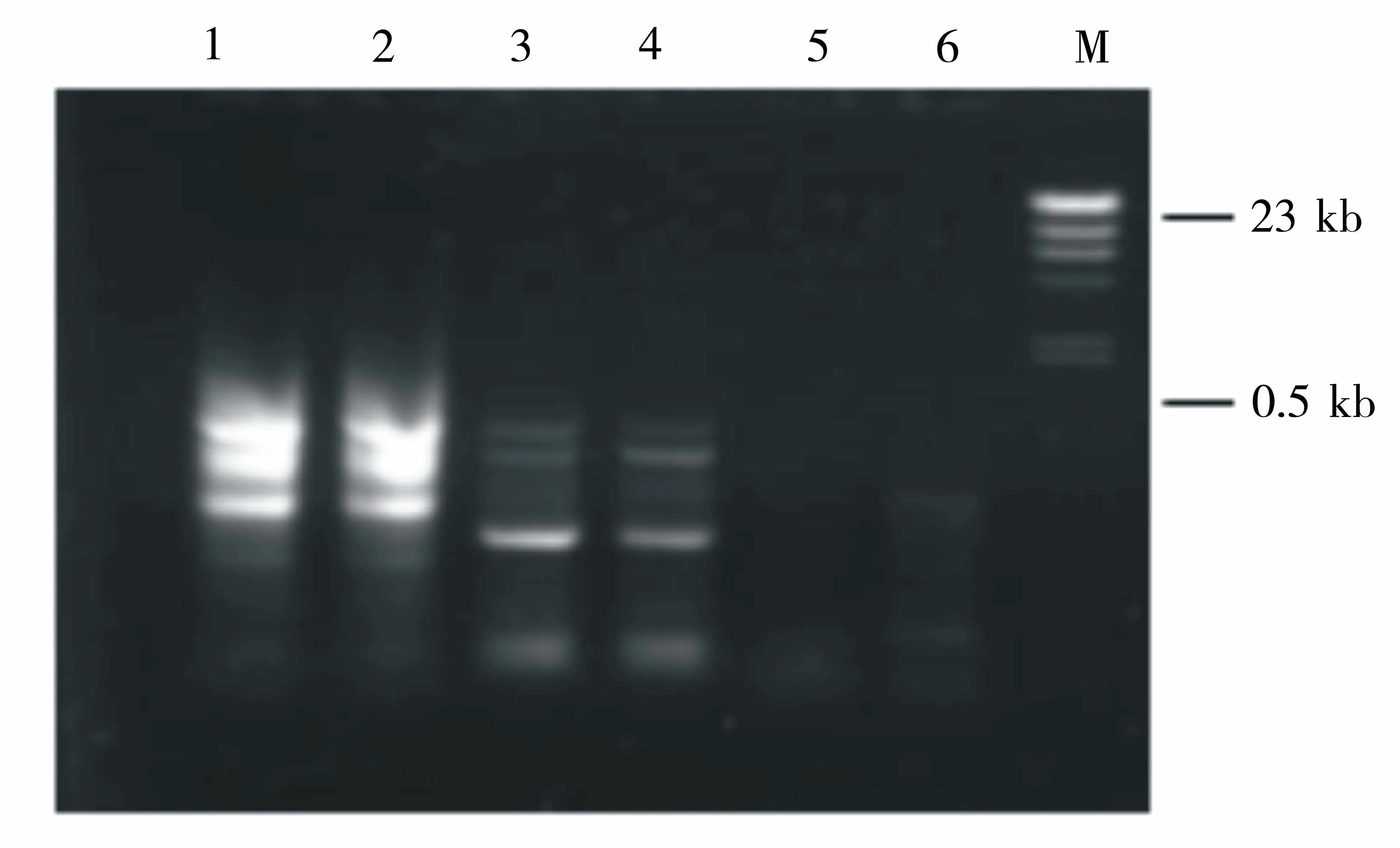
1.未经纯化的对照样品扩增; 2.未经纯化的盐激样品扩增; 3. 纯化过后的对照样品反转录后RAP-PCR; 4.纯化过后的盐激样品反转录后RAP-PCR; 5. 纯化过后的对照样品直接PCR;6. 纯化过后的盐激样品直接PCR; M:λDNA/Hind III图2 RNA纯化效果检测图Figure 2 Identification of purified and unpurified RNA samples by PCR and RT-PCR

M: 100 bp ladder;1~5:重扩增产物图3 差异片段经克隆测序后设计引物重扩增Figure 3 Reamplification of differentially expressed fragments
2.2 RNA的纯化效果检测
在进行差异显示试验时,一定要保证进行比较的一对RNA样品没有基因组DNA的污染,即PCR得到的带完全是反转录来的cDNA的结果。所以,对样品进行纯化效果的检测非常重要。图2显示的是RNA的纯化效果,由图2可知,纯化过后的RNA样品没有经过反转录直接作PCR,则在电泳时没有扩增信号,表明样品中的基因组DNA已被彻底去除;而经过反转录的样品的扩增结果条带清晰,则表明反转录效果良好,能满足进一步的试验要求。
2.3 RAP-PCR获得差异片段cDNA
所得到的总RNA样品经DNase I处理后,分别用11个碱基的反转录引物作反转录,得到的反转录产物再分别和10个PCR引物组合扩增,有100个引物组合。因为引物最初的设计是利用E.coli的基因组信息[6],而对于海洋细菌WP3,其引物是否合适还不得而知。采用WP3基因组DNA,对100个引物组合的扩增情况进行了筛选,发现其中的90个引物组合能得到足够多的带进行长胶电泳。因此,筛选出来的90对引物用于后面的差异显示研究。所有的90个引物组合共发现可能的差异表达片段60个,将这些带用无菌手术刀片切下,并记录好引物组合。
2.4 差异片段重扩增
因为差异片段在聚合的丙烯酰胺胶块里,而且是通过银染后从胶上挖下来的,所以在差异片段重扩增时,简单的直接做扩增或直接克隆测序,效果都不理想。笔者比较了其他回收丙烯酰胺胶块中核酸的方法,如4oC连续浸泡数天后去上清扩增等等,发现丙烯酰胺胶块中的核酸回收后在高温(95oC)短暂温浴(15~20 min)后的重扩增效果最为理想。
以胶带洗脱液为模板,扩增的引物按原来记录的引物组合,扩增条件跟原来相同。不能扩增出清晰条带或扩增结果不止一条带的全部丢弃,图3显示的是其中一部分的重扩增图。由图3可知,差异片段的大小大概在250~500 bp之间。重扩增后得到的带随后胶回收、克隆并测序。
2.5 反向Northern杂交反复验证差异基因
为了进一步验证高盐刺激下的基因表达,将用变性测序较得到的差异片段重新扩增纯化点到杂交膜上。运用反向Northern杂交方法对可能的差异基因进行进一步的确认。杂交实验显示,在从变性胶得到的40个片段中,只有4个片段给进一步确认是差异表达基因。反向Northern杂交的结果如图4。

A为正常对照;B为盐激样品;数字1、2、3、4代表差异表达的基因图4 反向Northern杂交筛选盐激(salt shock)中的差异表达基因Figure 4 Identification of salt shock regulated genes by reverse northern blot
2.6 差异片段的序列分析BLAST检索
经上海博亚生物公司测序后, 将测序结果进行BlastN、BlastP检索, 结果发现序列SS2与ShewanellahalifaxensisHAW-EB4中ferroxidase的同源性达到96%,序列SS3与ShewanellahalifaxensisHAW-EB4中gamma-butyrobetaine antiporter的同源性达到96%。而序列SS1、SS4在Genbank中找到的同源序列注释为hypothetical protein,其基因功能还有待进一步鉴定。差异表达基因的同源性分析见表2。

表2 高盐刺激下差异表达基因的同源分析Table 2 Homology analysis of nucleotide sequences of DNA fragments derived from RAP-PCR products of salt shock
3 讨论
微生物特别是细菌菌体里含有大量的RNase,因此要得到完整的RNA样品,就要最大可能地抑制RNase的活力。通过对几种典型的提取RNA的方法,发现MBI公司的TRI reagent方法简便,得到的RNA的质量较好。
差异显示技术在理论上简单易懂,在操作上简单、快速、灵敏、多用途,并可适用于微量起始即低丰度。然而,缺点是假阳性过高,为了克服这一缺点,一方面需进行重复试验,保证差异片段的可重复性,另一方面运用反向Northern杂交方法进行大量信息处理,最终得到差异表达基因。在原核细胞差异显示中,引物还是提高RNA 指纹图像重复性、降低差异条带假阳性的关键因素。因此,在进行引物设计时,始终强调引物特异性高与低的平衡,即既要保证全面覆盖mRNA 序列,又要防止与细胞内其他RNA 及DNA 的非特异性结合。同时还应尽量避免使用庞大数量的引物,因为从众多引物中筛选适宜的引物将是一份既耗精力,又耗财力的工作。尽管如此,对于基因组既不存在高频分散出现的重复序列,又没有高度重复的回文序列,设计任意引物用于原核细胞DDRT-PCR 不失为一种好方法。
因为抗逆农作物的培育需要,盐胁迫的研究在植物中研究最为广泛。微生物方面,已有很多模式微生物如E.coli、B.cillus等得到了比较系统的阐明。这些微生物对于盐胁迫的反应较为相似,主要是一些相容性分子如甜菜碱(glycine betaine)[7,8]及K+在细胞内的累积[9],在嗜压菌SS9的研究中也有发现相似的现象[10]。在本研究中也出现了类似情况,即SS3编码为gamma-butyrobetaine antiporter的基因上调。此外,还检测到了基因ferroxidase的下调,这可能暗示在对对数生长中期的WP3进行高盐浓度(12% NaCl)的刺激时,菌体的铁的吸收能力下降[11]。在对Synechocustissp.的功能基因组学研究中发现组氨酸激酶是该细菌遭遇盐胁迫时的感应器,但在本研究中组氨酸激酶的表达在盐激条件下并没有显著变化。本研究检测到的另外2个基因为hypothetical protein,这2个基因的功能还需要进一步的鉴定和功能确认。
[1]Vezzi A,Campanaro S,D'Angelo M,etal.Life at Depth:PhotobacteriumprofundumGenome Sequence and Expression Analysis[J].Science,2005,307:1459~1461.
[2]Deckert G,Warren PV,Gaasterland T,etal.The complete genome of the hyperthermophilic bacteriumAquifexaeolicus[J].Nature.1998,392:353~358.
[3]Benson N R,Wong R M,McClelland M.Analysis of the SOS response inSalmonellaentericaserovarTyphimuriumusing RNA fingerprinting by arbitrarily primed PCR[J].J Bacteriol,2000,182: 3490~3497.
[4]Bidle K A.Differential expression of genes influenced by changing salinity using RNA arbitrarily primed PCR in the archaeal halophileHaloferaxvolcanii[J].Extremophles,2003,7:1~7.
[5]Wang F,Wang P,Chen M,etal.Isolation of extremophiles with the detection and retrieval ofShewanellastrains in deep sea sediments from the West Pacific[J].Extremophiles,2004,8:165~168.
[6]Fislage R,Berceanu M,Humboldt Y,etal.Primer design for a prokaryotic differential display RT-PCR[J].Nucleic Acids Res,1997,25:1830~1835.
[7]Perroud B,Le Rudulier D.Glycine betaine transport inEscherichiacoli: osmotic modulation[J].J Bacteriol,1985,161:393~401.
[8]Glaasker E,Konings W N,Poolman B.Glycine betaine fluxes inLactobacillusplantarumduring osmostasis and hyper- and hypo-osmotic shock[J].J Biol Chem,1996,271:10060~10065.
[9]Kunin C M,Rudy J.Effect of NaCl-induced osmotic stress on intracellular concentrations of glycine betaine and potassium inEscherichiacoli,Enterococcusfaecalis,andstaphylococci[J].J Lab Clin Med,1991,118:217~224.
[10]Martin D D,Bartlett D H,Roberts M F.Solute accumulation in the deep-sea bacteriumPhotobacteriumprofundum[J].Extremophiles,2002,6:507~514.
[11]Attieh Z K,Mukhopadhyay C K,Seshadri V,etal.Ceruloplasmin ferroxidase activity stimulates cellular iron uptake by a trivalent cation-specific transport mechanism[J].J Biol Chem,1999,274:1116~1123.
2009-03-06
国家大洋开发协会项目(DY7000M-02)
李升康 (1975-),男, 湖北恩施人,理学博士,副教授,主要从事海洋微生物研究.
10.3969/j.issn.1673-1409(S).2009.01.018
Q786
A
1673-1409(2009)01-S064-04
