马家柚叶绿体基因组特征及其密码子偏好性分析
2024-06-15尹明华余璐周佳慧刘李娜徐文萱孙美龄
尹明华 余璐 周佳慧 刘李娜 徐文萱 孙美龄

DOI:10.13925/j.cnki.gsxb.20230558
摘 要:【目的】为了明确马家柚叶绿体基因组结构特征及其与同属类群的系统发育关系,阐明马家柚在柑橘属中的分类地位,对马家柚叶绿体基因组的特征及其密码子的偏好性进行分析。【方法】采用DNBSEQ-T7测序平台对马家柚进行测序,采用Noveplastys、CAP3、GeSeq和tRNAscan-SE软件对马家柚叶绿体基因组进行组装、注释;采用CGViewServer、MISA、REPuter、CodonW、Gview、IRscope、NADnaSP6.0软件对马家柚叶绿体基因组特征进行分析;采用MAFFT 7.0和FastTree 2.1.10软件对马家柚及其85个同科种和山小橘属3个外群种叶绿体基因组进行序列比对和建树。【结果】马家柚叶绿体基因组全长160 186 bp,包括1个大单拷贝(LSC)区、1个小单拷贝(SSC)区和2个反向重复(IR)区,为典型闭合环状双链结构。马家柚叶绿体基因组共注释到133个功能基因,包括88个编码蛋白(CDS)基因、8个核糖体RNA(rRNA)基因和37个转运RNA(tRNA)基因。马家柚叶绿体基因组共检测到79个简单序列重复(SSR)和34个长序列重复(Longrepeat)。马家柚叶绿体基因组非编码区的变异程度高于基因编码区,LSC区的变异性>SSC区>IR区,SC/IR边界较为保守。马家柚叶绿体基因组平均ENC值为48.02,密码子偏好性较弱。马家柚叶绿体基因组密码子使用偏好性主要受自然选择的影响,受内部突变的影响小。马家柚叶绿体基因有10个最优密码子(AAU、UGU、AAA、UUU、GCU、GGA、CCA、ACU、CGU、AGU),均以A、U结尾。马家柚与西双版纳东试早柚(KY055833,来源地:云南)、日本夏橙(ON193075,来源地:韩国)、福建六月早蜜柚(MT527726,来源地:福建)、福建琯溪蜜柚(MN782007,来源地:福建)有亲缘关系。【结论】马家柚是一个柑橘属中较为独特的品种,该研究结果为进一步研究马家柚的遗传资源、物种资源鉴定和系统发育分析提供了理论依据。
关键词:马家柚;叶绿体基因组;序列特征;密码子偏好性;最优密码子;系统发育分析
中图分类号:S666.3 文献标志码:A 文章编号:1009-9980(2024)05-0824-23
收稿日期:2023-12-29 接受日期:2024-03-14
基金项目:国家自然科学基金项目(31960079、31860084、32060092);江西省现代农业产业技术体系建设专项(JXARS-13-赣东站);2022年上饶市科技专项项目(饶科发[2023]5号社发类)(2022A008));江西省科技厅重点研发计划一般项目(20202BBG73010);江西省教育厅科学技术研究项目(GJJ201704、GJJ211729);上饶市科技局平台载体建设项目(2020J001)
作者简介:尹明华,女,教授,主要从事植物生物技术方面的研究。Tel:0793-8153721,E-mail:yinminghua04@163.com
Analysis of the chloroplast genome sequence characteristics and its code usage bias of Citrus maxima (L.) Osbeck ‘Majiayou
YIN Minghua1, 2, 3, 4, 5, YU Lu1, ZHOU Jiahui1, LIU Lina1, XU Wenxuan1, SUN Meiling1
(1College of Life Sciences, Shangrao Normal University, Shangrao 334001, Jiangxi China; 2Shangrao Agricultural Technology Innovation Research Institute, Shangrao 334001, Jiangxi China; 3Majiayou Industry Research Institute of Shangrao Normal University, Shangrao 334001, Jiangxi China; 4Key Laboratory of Protection and Utilization of Medicinal and Edible Plant Resources in Shangrao City, Shangrao 334001, Jiangxi China; 5Key Laboratory of Germplasm Conservation and Utilization of Potato and Taro Crops in Shangrao City, Shangrao 334001, Jiangxi China)
Abstract: 【Objective】 Citrus maxima (L.) Osbeck ‘Majiayou was approved by the former Ministry of Agriculture as a national geographical indication agricultural product in 2010. At present, all counties and cities in Shangrao City are vigorously developing the C. maxima (L.) Osbeck ‘Majiayou industry. It is urgent to trace the origin of C. maxima (L.) Osbeck ‘Majiayou to ensure its authenticity. There have been some studies indicating that the genetic relationship between C. maxima (L.) Osbeck ‘Majiayou and C. maxima (L.) Osbeck ‘Xinmuyou in the surrounding areas is relatively close, and it is quite likely that C. maxima (L.) Osbeck ‘Majiayou is a variant strain derived from the bidirectional (natural and artificial) selection of local pomelo. However, the above research has not yet solved the phylogenetic problem of C. maxima (L.) Osbeck ‘Majiayou. The study aimed to rectify the source of C. maxima (L.) Osbeck ‘Majiayou and explore the phylogenetic relationship with other Citrus plants through surveying the characteristics of the chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou and its codon preference. 【Methods】 The total DNA extraction from the leaves of C. maxima (L.) Osbeck ‘Majiayou was performed using an improved CTAB method. The purity of the DNA was detected using the NanoDrop 2000 spectrophotometer method; Preliminary quantification of the DNA library using Invitrogen Qubit? 2.0 fluorescence quantitative instrument method; The detection of inserted fragments in the DNA library was carried out using the Agilent 2100 biological analyzer system; The accurate quantification of the effective concentration in the DNA library was carried out using real-time fluorescence quantitative PCR method; The DNA library was sequenced using the DNBSEQ-T7 sequencer method. The assembly of the chloroplast genome was carried out using Noveplastys and CAP3 software; The annotation of the chloroplast genome was performed using GeSeq and tRNAscan-SE software; The production of the chloroplast genome map was carried out using OGDRAW software. The analysis and statistics of GC content in the large single copy region (LSC), small single copy region (SSC), and reverse repeat region (IR) of the chloroplast genome were conducted using CGViewServer software; The SSR analysis of the chloroplast genome was performed using MISA software; The Longrepeat analysis of the chloroplast genome was performed using REPuter software; The calculation and analysis of the RSCU of the chloroplast genome were carried out using CodonW software; The drawing of chloroplast genome variation circles and the calculation of sequence similarity for C. maxima (L.) Osbeck ‘Majiayou and its 18 congeneric species were performed using Gview software; The mapping of IR structural variations in chloroplast genomes of C. maxima (L.) Osbeck ‘Majiayou and its 18 congeneric species was performed using IRscope software; The calculation of the chloroplast genome Pi of C. maxima (L.) Osbeck ‘Majiayou and its 18 congeneric species was carried out using NADnaSP6.0 software; The sequence alignment and tree construction of chloroplast genomes of C. maxima (L.) Osbeck ‘Majiayou and its 85 same family species, as well as three outer groups of Glycosmis, were carried out using MAFFT 7.0 software and FastTree 2.1.10 software, respectively. 【Results】 The chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou had a total length of 160 186 bp, including 1 LSC region (87 791 bp), 1 SSC region (18 395 bp), and 2 IR regions (including IRa and IRb, both 27 000 bp). Its structure presented a typical closed circular double stranded structure. The average GC content of the chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou was 38.47%, with the GC content in the IR region being higher than that in the LSC and SSC regions. The chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou annotated 133 functional genes, including 88 coding sequence (CDS) genes, 8 ribosomal RNA (rRNA) genes, and 37 transporter RNA (tRNA) genes. A total of 79 simple repeat sequences (SSRs) were detected in the chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou, including only single nucleotide repeat sequences and trinucleotide repeat sequences. The single nucleotide repeat sequences were mostly A and T repeats. A total of 34 long repeat sequences were detected in the chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou, including 13 dispersed repeat D (1739-135 819 bp) and 21 palindrome repeat P (421-125 236 bp). The chloroplast genome sequences of C. maxima (L.) Osbeck ‘Majiayou and its 18 congeneric species were highly conserved, with significant sequence differences between genes such as petN, petL, psbI, psbK, psaI, pafII, trnT-GGU, trnR-UCU, trns-GGA, and trnL-UAA in the LSC and SSC regions. The variation ranges of the nucleotide diversity in the chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou was from 0 to 0.00629; The degree of variation in the non coding region of the chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou was higher than that in the gene coding region. The overall variability was higher in the LSC region, followed by the SSC region. The IR region had the lowest variability and was the most conservative region; The SC/IR boundaries of the chloroplast genomes of C. maxima (L.) Osbeck ‘Majiayou and its 18 congeneric species were relatively conservative. The bias analysis of synonymous codons showed that the variation trend of GC content at three positions of the chloroplast genome codon of C. maxima (L.) Osbeck ‘Majiayou and its 18 related species was GC3<GC2<GC1, with an ENC value ranging from 26.309 to 61 and an average of 48.04. The codon bias was weak, and all codons except UGG, UUG, and AUG ended in A and U. Neutral plot analysis showed that the GC3 and GC12 contents of the chloroplast genes of C. maxima (L.) Osbeck ‘Majiayou and its 18 congeneric species were mostly distributed above the diagonal, with an internal mutation contribution rate of only 2.5% and a natural selection contribution rate of 97.5%. The codon usage bias of the chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou and its 18 congeneric species was mainly influenced by the natural selection, and was less affected by internal mutation pressure. The ENC plot analysis showed that there were significant differences between the actual and expected values of most of the genes ENC in the chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou and its 18 congeneric species, and the distribution of GC3 values was relatively concentrated, indicating that natural selection was an important factor affecting the codon usage bias of chloroplast genome. The PR2 plot analysis showed that the chloroplast genomes of C. maxima (L.) Osbeck ‘Majiayou and its 18 congeneric species exhibited C>G and T>A phenomena at the third synonymous codon position, indicating that the codon usage preference of C. maxima (L.) Osbeck ‘Majiayou was influenced not only by internal mutations but also by natural selection. There were a total of 10 optimal codons in the chloroplast genome of C. maxima (L.) Osbeck ‘Majiayou, including AAU, UGU, AAA, UUU, GCU, GGA, CCA, ACU, CGU, and AGU, all ending in A and U. C. maxima (L.) Osbeck ‘Majiayou was closely related to C. maxima (Dongshizaoyou in Xishuangbanna, KY055833, source: Yunnan), Japanese summer orange (C. natsudaidai, ON193075, source: South Korea), C. maxima ‘Liuyuezao (MT527726, source: Fujian), and C. maxima (Burm.) Merr. ‘Guanximiyou (MN782007, source: Fujian). 【Conclusion】 C. maxima (L.) Osbeck ‘Majiayou is a relatively unique variety in the Citrus genus. The research results would provide a theoretical basis for further research on the genetic resources, species identification, and phylogenetic analysis of C. maxima (L.) Osbeck ‘Majiayou.
Key words: Citrus maxima (L.) Osbeck ‘Majiayou; Chloroplast genome; Sequence characteristics; Codon usage bias; Optimal codons; Phylogenetic analysis
尹明华
马家柚[Citrus maxima (L.) Osbeck ‘Majia-
you]为芸香科(Rutaceae)柑橘亚科(Aurantioideae)柑橘属(Citrus)亚热带常绿果树,为江西省地方特色红心柚品种,原产于江西省上饶市广丰区大南镇古村马家村[1],是由当地农业部门通过对诸多柑橘资源进行多年筛选和普查而获得的优良柚种质,2009年由江西省农作物品种审定委员会审定并命名,2010年被农业部核准为国家地理标志农产品[2]。马家柚药食兼用。食用,马家柚果大皮黄,肉粉多汁,甜酸清爽,口味独特,营养丰富[3];药用,马家柚的柚皮、柚肉、柚花和柚核含有多种活性成分如β-柠檬烯、黄酮、柠檬苦素等,具有理气和胃、消食化痰、镇痛消炎、抗癌抑瘤、抗病毒、抗氧化、解酒毒等功效[4]。目前对马家柚的研究主要集中于套袋处理[3,5-6]、营养成分[7]、光合特性[4]、花粉直感[2]、授粉昆虫[1]、发酵工艺[8]、解剖观察[9]、土壤养分[10]等方面,但马家柚叶绿体基因组特征及其密码子偏好性分析的研究尚未见报道,马家柚的进化起源及其系统发育的亲缘关系尚未得到明确的鉴定。目前,上饶市各县市均在大力发展马家柚产业,如何保证广丰马家柚的道地性,急需对广丰马家柚正本溯源。
通过对同属植物叶绿体基因组序列的比较和分析,构建同属植物系统发育树,可综合评估该品种的系统发育位置和演化关系[11]。叶绿体是绿色植物细胞内可将光能转化为化学能的半自主性细胞器[12],其基因组为四分体双链环状结构,一般由1个大单拷贝(Large single copy,LSC)区、1个小单拷贝(Small single copy,SSC)区和2个反向重复(Inverted repeats,IRs,包括IRa和IRb)区组成[13]。叶绿体编码区的核酸替代速率相对较低的特点为植物深层次系统进化研究提供了必要条件。越来越多的叶绿体编码基因被广泛应用于不同科、目乃至整个被子植物的系统学研究,对植物间的系统发育研究和进化关系分析做出了重大贡献[14]。在植物系统学研究中较为常用的叶绿体编码基因有rbc、matK、atpB和ndhF等[15]。叶绿体基因组序列揭示了植物物种内部和物种之间的序列差异和结构变异较大,这些信息对了解重要作物的适应能力,促进密切相关物种的育种以及识别和保护有价值的性状具有重大的意义[16]。通过完整叶绿体基因组的多态性和高通量基因组比较,对复杂的遗传关系进行探究,目前已从属传递到科,并达到目水平[17]。叶绿体的完整序列也为分子育种和DNA条形码标记的开发提供了有用的信息,并已在植物种质资源的保护方面得到了有效的应用。叶绿体基因组包含的简单重复序列(Simple sequence repeat,SSR)和长重复序列(Long repeat sequence,Longrepeat)均可以作为有效的DNA分子标记用于物种遗传多样性和遗传稳定性的检测,有利于植物的分子辅助育种和种质资源保存[18-20]。徐世荣等[21]对六月早蜜柚(C. maxima ‘Liuyuezao)的叶绿体基因组及其特征进行了分析,发现六月早蜜柚与甜橙(C. sinensis)、柠檬(C. limon)和C. platymamma的亲缘关系较近;Xu等[22]对福建琯溪蜜柚[C. maxima (Burm.) Merr. ‘Guanximiyou]的叶绿体基因组序列进行了分析,发现福建琯溪蜜柚与柚(C. maxima)、甜橙(C. sinensis)、C. platymoma和柠檬(C. limon)亲缘关系较近;Zhang等[23]对云南红河柑橘(C. hongheensis)的叶绿体基因组特征进行了分析,发现云南红河柑橘与C. maxima亲缘关系较近;Cai等[24]对手指柠檬(C. australasic)栽培种的叶绿体基因组序列进行了分析,发现手指柠檬栽培种与C. medica亲缘关系较近;Su等[25]对手指柠檬(C. australasic)栽培种的叶绿体基因组序列进行了分析,确定了阿曼酸橙(Omani lime,C. aurantiifolia)3个基因间区域和94个简单序列重复(SSR),是具有种间关系分辨率的潜在信息标记,可以利用这些标记更好地了解栽培柑橘的起源。Ishikawa等[26]利用叶绿体全基因组序列及其生物多样性对扁实柠檬(Shiikuwasha,C. depressa Hayata)多个谱系进行评价,发现与野生种群相比,栽培种群已失去基因的多样性。Bausher等[27]通过组织与系统发育以及与其他被子植物的关系对C. sinensis (L.) Osbeck ‘Ridge Pineapple的叶绿体基因组序列进行了分析,发现反向重复区的扩展包括rps19和部分rpl22以及rpl22的两个截短拷贝的存在是不寻常的,完整的柑橘叶绿体基因组序列的可用性可为叶绿体基因工程提供关于基因间隔区和内源性调控序列有价值的信息。目前,有关广丰马家柚的叶绿体基因组组装、注释、基因组特征及系统发育方面的研究尚未见报道。笔者在本研究中对广丰马家柚叶绿体基因组序列组装注释,明确其叶绿体基因组特征、密码子偏好性及系统进化等相关问题,为广丰马家柚种质资源的鉴定、开发和利用提供参考。研究首次对广丰马家柚的叶绿体基因组进行测序、组装和注释,进一步分析其叶绿体基因组特征和密码子偏好性等,筛选有效的最优密码子,并将其与已公布叶绿体基因组的柑橘属同属种构建系统发育树,阐明广丰马家柚与其他柑橘属同属种的进化关系及其在系统发育中的地位,可为柑橘属植物的遗传进化研究提供思路,也为种质资源开发利用和叶绿体基因工程研究提供参考。
1 材料和方法
1.1 材料
广丰马家柚(代号:MJY)盆栽苗由上饶师范学院马家柚产业研究院提供。
1.2 方法
1.2.1 DNA提取和测序 马家柚叶片的总DNA提取采用改良的CTAB法[28];叶片DNA纯度的检测采用NanoDrop 2000分光光度计(美国,Thermo Scientific公司)检测;马家柚叶片DNA文库采用美国Invitrogen Qubit? 2.0荧光定量仪初步定量;马家柚叶片DNA文库插入片段采用Agilent 2100生物分析仪系统检测;马家柚叶片DNA文库有效浓度采用实时荧光定量PCR(Real-time quantitative PCR,RT-qPCR)准确定量;马家柚叶片DNA文库采用广州佰数生物科技有限公司(Bio&Data Biotechnologies)DNBSEQ-T7测序仪平台(华大智造)测序。
1.2.2 叶绿体全基因组的组装与注释 采用fastp V0.23.2软件(默认参数)[29]过滤马家柚叶片DNA文库Raw Data原始数据,去除马家柚叶片DNA文库低质量Reads,获得马家柚叶片DNA文库高质量Clean Data。马家柚叶绿体基因组的组装采用Noveplastys[30]和CAP3[31]软件,Noveplastys和CAP3软件适用于组装,其中Noveplastys是主程序,在Noveplastys未环化情况下,CAP3软件参与序列环化处理;马家柚叶绿体基因组的注释采用GeSeq[32]和tRNAscan-SE[33]软件,GeSeq和tRNAscan-SE软件用于基因组注释,tRNA-scan-SE用于补充GeSeq对tRNA注释的不足;马家柚叶绿体基因组图谱的制作采用OGDRAW[34]软件。注释完成后,将马家柚叶绿体基因组序列提交至NCBI(https://www.ncbi.nlm.nih.gov/),获得登录号(PP024602)。
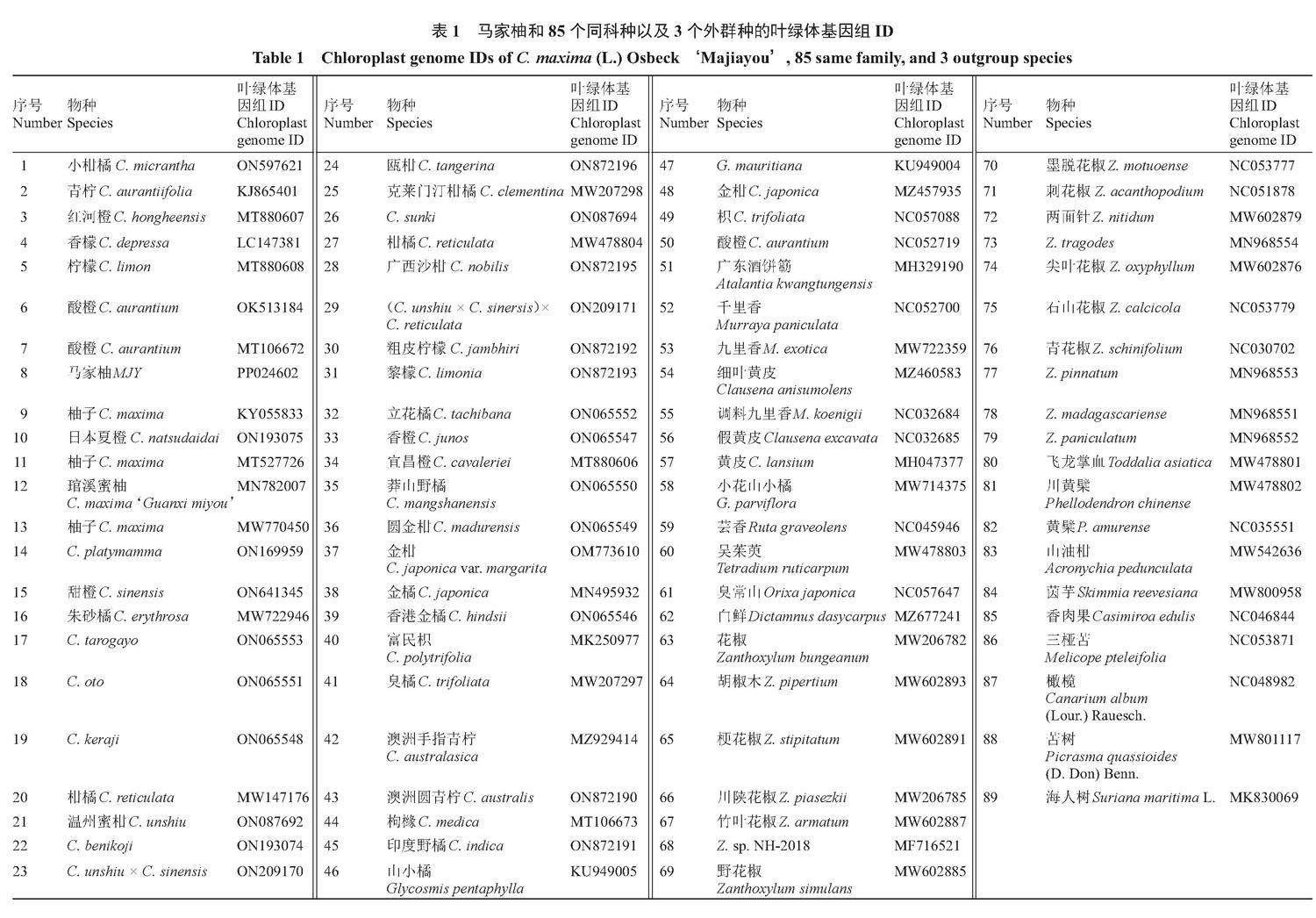
1.2.3 叶绿体基因组特征分析 马家柚叶绿体基因组的大单拷贝区(LSC)、小单拷贝区(SSC)和反向重复区(IR)GC含量的分析和统计采用CGViewServer[35]软件;马家柚叶绿体基因组的SSR分析采用MISA(MIcroSAtellite identification tool)[36]软件;马家柚叶绿体基因组的Longrepeat分析采用REPuter[37]软件;马家柚叶绿体基因组同义密码子相对使用度(relative synonymous codon usage,RSCU)的计算和分析采用CodonW软件[38];马家柚及其18个同属种(表1序号1~19)叶绿体基因组变异圈图的绘制和序列相似性的计算均采用Gview[39]软件;马家柚及其18个同属种(表1序号1~19)叶绿体基因组IR结构变异的绘图采用IRscope[40]软件;马家柚及其18个同属种(表1序号1~19)叶绿体基因组核苷酸多态性(Nucleotide polymorphism,Pi)的计算采用NADnaSP6.0[41]软件;马家柚及其85个同科种(芸香科柑橘亚科、芸香亚科和飞龙掌血亚科)和3个外群种(橄榄NC048982、苦树MW801117、海人树MK830069)(表1)叶绿体基因组的序列比对和建树分别采用MAFFT 7.0[42]软件和FastTree 2.1.10[43]软件。
1.2.4 叶绿体基因组密码子使用偏好性分析 GC3-GC12(Neutrality-plot)、ENC-plot、PR2-plot和最优密码子分别采用参考Liu等[19]的方法进行分析。其中,GC3-GC12分析用R语言做GC3和GC12的线性回归分析;ENC-plot用EMBOSS(6.6.0.0)计算ENC,用R语言绘制ENC-plot;PR2-plotPR2通过A3、T3、G3、C3(分别表示密码子第三位碱基的A、T、G、C含量)分别计算AT-bias[A3/(A3+T3)]与GC-bias[G3/(G3+C3)],利用二者的对应关系来分析选择和突变对密码子使用模式的影响,所用数据由字写python脚本计算。
1.2.5 叶绿体基因组特异性标记筛选 通过分析,马家柚叶绿体基因组与KJ865401、KY055833、LC147381、MK250977、MN495932、MT106672、MT106673、MT880606、MT880607、MT880608、MW147176、MW207297、MW207298、MW478804、MW722946、MW770450、MZ929414、OK513184、OM773610、ON065546、ON065547、ON065548、ON065549、ON065550、ON065551、ON065552、ON065553、ON087692、ON087694、ON169959、ON193074、ON209170、ON209171、ON597621、ON641345、ON872190、ON872191、ON872192、ON872193、ON872195、ON872196存在变异的序列,通过这些变异的序列筛选出马家柚叶绿体基因组的特异性标记。
2 结果与分析
2.1 马家柚叶绿体基因组的基本结构
马家柚叶绿体基因组全长160 186 bp,包括1个LSC区(87 791 bp)、1个SSC区(18 395 bp)和2个IR区(包括IRa和IRb,均为27 000 bp),其结构呈典型闭合环状双链结构(图1)。马家柚叶绿体基因组平均GC含量为38.74%,其中IR区的GC含量(42.95%)高于LSC区(36.8%)和SSC区(33.34%)。
2.2 马家柚叶绿体基因组的基因组成
马家柚叶绿体基因组共注释到133个功能基因(表2),包括88个编码蛋白(Coding sequence,CDS)基因、8个核糖体RNA(rRNA)基因和37个转运RNA(tRNA)基因,其中假基因为0个。按照基因功能,马家柚叶绿体基因组的基因可分为四大类:第一类是与光合作用有关的44种基因;第二类是自我复制的77种基因;第三类是其他功能的6种基因;第四类是未知功能的6种基因。其中,trnH-GUG、trnK-UUU、trnI-GAU、trnA-UGC、trnG-UCC、trnV-UAC、trnL-UAA、rpoC1、ndhB、ndhA、rpl2、rpl16、petB、atpF、petD、rps16、rps12基因具有2个外显子,rps12、clpP1、pafI基因具有3个外显子(rps12有2个拷贝,每个拷贝具有3个外显子,且两个拷贝共享第一个外显子,第一个外显子位于LSC区域,另外两个外显子位于IR区域);完全在LSC区的基因有80个(21个tRNA基因和59个CDS基因),完全在SSC区的基因有11个(1个tRNA基因和10个CDS基因),完全在IRB和IRA区的基因有18个(4个rRNA基因、7个tRNA基因和7个CDS基因),在SSC-IRB连接处的基因有2个CDS基因(ndhF和ycf1),在LSC-IRB连接处的基因有1个tRNA基因(trnH-GUG),在SSC-IRA连接处的基因有1个CDS基因(ycf1)。
2.3 马家柚叶绿体基因组SSR和Longrepeat分析
马家柚叶绿体基因组共检测到79个SSR(表3),只包括单核苷酸重复序列和三核苷酸重复序列,单核苷酸重复序列有78个(97.44%),其中77个为A和T重复(98.72%),1个为G和C重复(1.28%);三核苷酸重复序列1个(1.27%)。马家柚18个同属种叶绿体基因组(KJ865401、KY055833、LC147381、MN782007、MT106672、MT527726、MT880607、MT880608、MW722946、MW770450、OK513184、ON065548、ON065551、ON065553、ON169959、ON193075、ON597621、ON641345)分别具有77、76、84、79、80、79、76、79、76、76、80、76、76、76、76、79、77和76个SSR,且均以A和T的单核苷酸重复序列为主,说明马家柚及其18个同属种叶绿体基因组SSR偏好使用A和T碱基。马家柚叶绿体基因组共检测到34个Longrepeat(表4),包括分散重复(Dispersed repeats,D)和回文重复(palindromic repeats,P),其中分散重复D分为正向重复(forward repeats,F)、反向重复R(reverse repeats,R)和互补重复C(complement repeats,C)3种。马家柚叶绿体基因组具有13个分散重复D(30~50 bp)和21个回文重复P(30~27 000 bp)。
2.4 叶绿体基因组特异性标记筛选
与KJ865401、KY055833、LC147381、MK250977、MN495932、MT106672、MT106673、MT880606、MT880607、MT880608、MW147176、MW207297、MW207298、MW478804、MW722946、MW770450、MZ929414、OK513184、OM773610、ON065546、ON065547、ON065548、ON065549、ON065550、ON065551、ON065552、ON065553、ON087692、ON087694、ON169959、ON193074、ON209170、ON209171、ON597621、ON641345、ON872190、ON872191、ON872192、ON872193、ON872195、ON872196比较,马家柚叶绿体基因组的部分特异性标记见表5。马家柚叶绿体基因组的rps18、rpl36、psbZ、psbJ、psbF基因有1个变异位点,rps7、rpl23、rpl14、psbK、psbH、psaC、psaJ、atpE、ndhB、petL、pafI、petN、ndhE基因有2个变异位点,atpH基因有3个变异位点,rpl32、rpl33、psbL、ndhC、petA、ndhK、petD基因有4个变异位点,rps11、rpl16、clpP1基因有5个变异位点,rps15、rps12、ndhI、atpB基因有6个变异位点,ndhJ、rps4、rps2、rps16、infA基因有7个变异位点,ndhG、cemA基因有8个变异位点,rps8、psbD、atpI、petB基因有9个变异位点,rpl20、pafII基因有10个变异位点,ndhA基因有11个变异位点,atpF、psaB、psbA基因有12个变异位点,rps19、rps14、rpoA、psbB基因有14个变异位点,rps3、psbC、atpA基因有15个变异位点,ndhH基因有16个变异位点,psaA基因有19个变异位点,accD基因有23个变异位点,rpoC1基因有25个变异位点,rbcL基因有28个变异位点,ccsA基因有29个变异位点,rpl22基因有34个变异位点,ndhD基因有36个变异位点,matK基因有42个变异位点,rpoB基因有44个变异位点,ycf2基因有52个变异位点,rpoC2基因有69个变异位点,ndhF基因有71个变异位点,ycf1基因有254个变异位点。
2.5 叶绿体基因组比对分析
马家柚及其18个同属种叶绿体基因组的变异圈图(图2)、mVIST结构变异图(图3)和Pi多样性指数分析图(图4)表明,马家柚及其18个同属种的叶绿体基因组序列高度保守,LSC和SSC区中petN、petL、psbI、psbK、psaI、pafII、trnT-GGU、trnR-UCU、trns-GGA、trnL-UAA等基因之间存在较大的序列差异。由图4可知,马家柚叶绿体基因组核苷酸多样性的变化范围为0~0.006 29;马家柚叶绿体基因组非编码区的变异程度高于基因编码区,LSC区的变异性整体较高,其次是SSC区,IR区变异性最低,为最为保守的区域;通过Pi(≥0.003 6)筛选出10个高变异区域,均位于LSC和SSC区,LSC区有8个高变异区域:Inter、trnS-GCU_trnG-UCC、trnT-UGU_trnL-UAA、accD-psaI、psbE-petL、rps18、rps3-rpl22、rpl22;SSC区有2个高变异区域:rpl32_trnL-UAG、ycf1-2。
2.6 叶绿体基因组的SC/IR/边界分析
马家柚及其18个同属种叶绿体基因组四分体结构的SC/IR边界收缩扩张情况(图5)表明:马家柚及其18个同属种叶绿体基因组的4个边界较为保守。LSC/IRb[LSC和IRb之间的结合点(junction sites),JLB]边界均位于rps19基因内,rps19基因在IRb区内长为46 bp,在LSC区内长为233 bp;IRb/SSC(SSC和IRb之间的结合点,JSB)边界均位于IRb区的ycf1基因和SSC区的ndhF基因之间,ycf1基因距离IRb/SSC边界均为2 bp。SSC/IR(SSC和IRa之间的结合点,JSA)边界均位于ycf1基因内,ycf1基因在SSC区长18 264~18 283 bp,ycf1基因在IRa区长5607~5622 bp。IRa/LSC(LSC和IRa之间的结合点,JLA)边界均位于IRa区的rpl2基因和LSC区的trnH基因之间,trnH基因距离IRb/SSC边界均为1 bp。
2.7 马家柚叶绿体基因组密码子使用偏好性分析
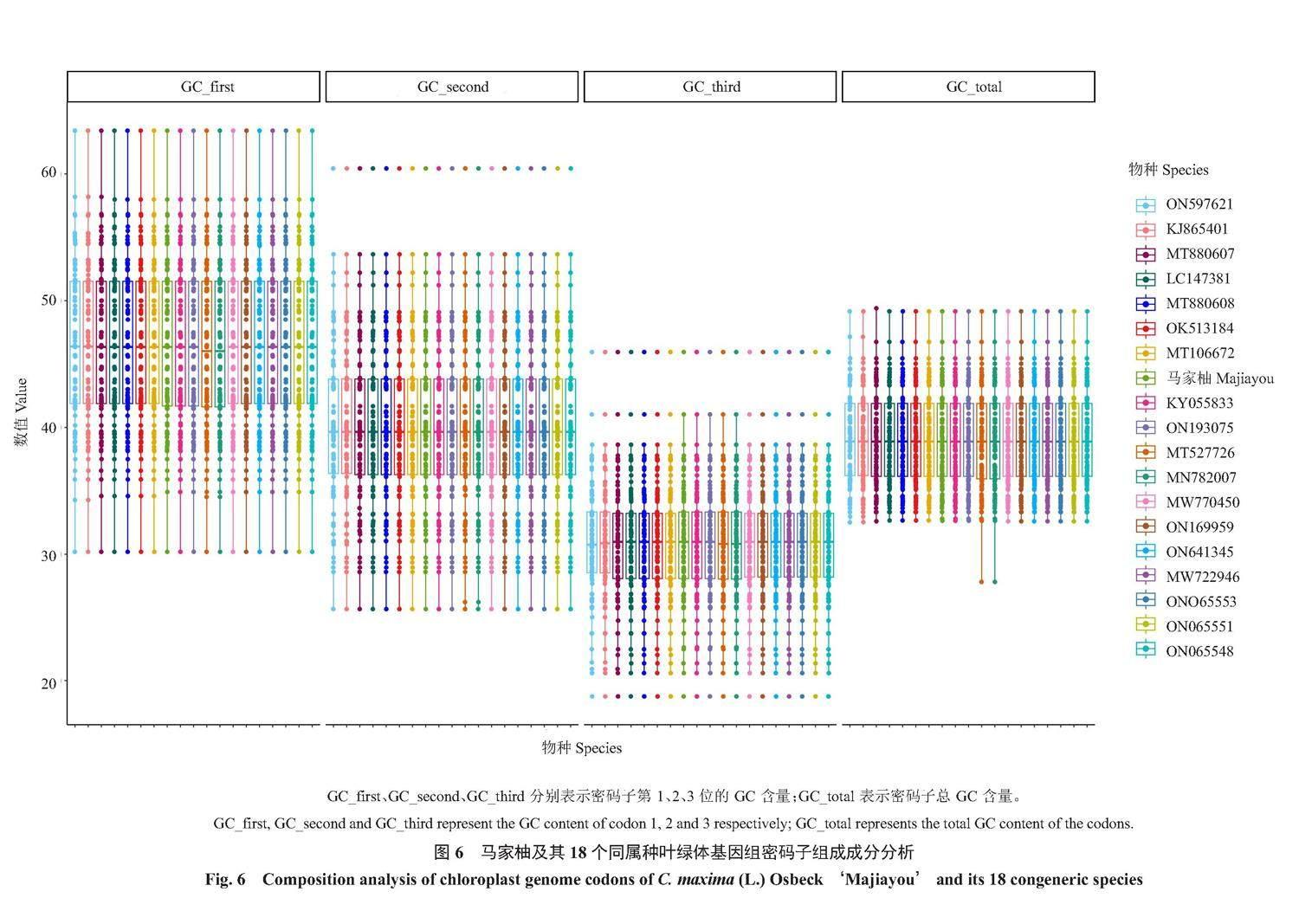
2.7.1 同义密码子的偏好性分析 马家柚及其18个同属种叶绿体基因组CDS基因密码子3个位置GC含量的平均值为39.06%,GC1、GC2、GC3含量分别为46.37%、40.02%、30.79%,马家柚及其18个同属种叶绿体基因组密码子3个位置GC含量的变化趋势为:GC3<GC2<GC1(图6);马家柚叶绿体基因组88个CDS基因密码子的ENC值介于28.311~61.000之间,平均值为48.02,84个基因的ENC值>35,4个基因的ENC值<35,密码子偏好性较弱;马家柚18个同属种叶绿体基因组CDS基因密码子的ENC值介于26.309~61.000之间,平均值为48.04,1511个基因的ENC值>35,71个基因的ENC值<35,密码子偏好性同样较弱。马家柚叶绿体基因组88个CDS基因序列共有32个RSCU>1的密码子,在这32个密码子中,除UGG、UUG、AUG外,其余都以A、U结尾;马家柚18个同属种叶绿体基因组CDS基因序列共有576个RSCU>1的密码子,在这576个密码子中,除UGG、UUG、AUG外,其余也均以A、U结尾,密码子同样偏好以A、U结尾(图7)。
2.7.2 中性绘图分析(GC3-GC12分析)、ENC-plot分析和PR2-plot分析 马家柚及其18个同属叶绿体基因的GC3含量分布在0.187 5~0.459 5之间,GC12含量分布在0.256 4~0.634 1之间,二者绝大多数沿对角线上方分布(图8)。两者的线性相关系数r=0.031 6(R2=0.001),相关显著(p<0.05),说明GC12与GC3相关性不显著。回归系数为0.025,内部突变贡献率仅2.5%,自然选择贡献率为97.5%,表明马家柚及其18个同属种叶绿体基因组密码子使用偏好性主要受自然选择的影响,而受内部突变压力的影响小。马家柚及其18个同属种叶绿体基因组大部分基因位于标准曲线的下方(图8),大部分基因ENC的实际值与预期值存在较大差异,且GC3值分布较为集中,可见自然选择是影响马家柚及其18个同属种叶绿体基因组密码子使用偏好性的重要因子。从G3/GC3轴看,马家柚及其18个同属种叶绿体基因组中较多的基因分布于PR2-plot图的下部分(图8),说明4种碱基在同义密码子第3位上存在C>G现象。从A3/AU3轴看,马家柚及其18个同属种叶绿体基因组中较多的基因分布于PR2-plot图的左部(图8),说明4种碱基在同义密码子第3位上存在T>A现象。如密码子使用存在偏好性只受内部突变压力影响时,C和G以及A和T在第3位上的分布应相等,说明马家柚及其18个同属种叶绿体基因组密码子使用偏好性在受内部突变影响的同时也会受到自然选择的影响。
2.7.3 最优密码子确定 马家柚叶绿体基因组满足相对同义密码子使用度(Relative synonymous codon usage,RSCU)>1(高频率密码子)且ΔRSCU(=RSCU高表达-RSCU低表达)≥0.08的最优密码子有AAU、UGU、AAA、UUU、GCU、GGA、CCA、ACU、CGU、AGU(表6),共10个,均以A、U结尾。说明马家柚叶绿体基因组密码子偏好性是以A和U结尾。
2.8 系统发育分析
基于马家柚及其85个同科种(芸香科柑橘亚科、芸香亚科和飞龙掌血亚科)和3个外群种(橄榄NC048982、苦树MW801117、海人树MK830069)叶绿体基因组构建的系统发育树(图9)表明,马家柚[MJY,C. maxima (L.) Osbeck ‘Majiayou]与柚子(C. maxima,KY055833)、日本夏橙(C. natsudaidai,ON193075)、柚子(C. maxima,MT527726)、柚子(C. maxima,MN782007)单独聚为一分支。在这个分支中,马家柚[MJY,C. maxima (L.) Osbeck ‘Majiayou]又单独成一分支。说明马家柚[MJY,C. maxima (L.) Osbeck ‘Majiayou]与西双版纳东试早柚(C. maxima,KY055833,来源地:云南)、日本夏橙(C. natsudaidai,ON193075,来源地:韩国)、福建六月早蜜柚(C. maxima ‘Liuyuezao,MT527726,来源地:福建)、福建琯溪蜜柚[C. maxima (Burm.) Merr. ‘Guanximiyou,MN782007,来源地:福建]有亲缘关系,是一个柑橘属中较为独特的品种。
3 讨 论
马家柚叶绿体基因组全长160 186 bp,与其他柑橘属植物叶绿体基因组大小差别不大,伍泓昆[44]的研究表明,红河大翼橙、枳、金柑、莽山野橘、柚和枸橼叶绿体基因组全长分别为161 419 bp、160 589 bp、160 184 bp、160 152 bp、160 097 bp和159 952 bp;徐世荣等[21]和Xu等[22]的研究也表明,福建六月早蜜柚(C. maxima ‘Liuyuezao)和福建琯溪蜜柚[C. maxima (Burm.) Merr. ‘Guanximiyou]的叶绿体基因组全长均为160 186 bp,与马家柚叶绿体基因组全长相同,说明在柑橘属内各个种的叶绿体基因组相对保守。福建六月早蜜柚[21]、福建琯溪蜜柚[22]、云南红河柑橘(C. hongheensis)[23]、阿曼酸橙(Omani lime,C. aurantiifolia)[25]、扁实柠檬(Shiikuwasha,C. depressa Hayata)[26]、C. sinensis (L.) Osbeck ‘Ridge Pineapple[27]、手指柠檬(C. australasic)栽培种[24]和马家柚(MJY)叶绿体基因组平均GC含量为38%~39%,其中IR区的GC含量高于LSC区和SSC区,SSC区的GC含量最低,究其原因,IR区的rRNA基因的GC含量高,而SSC区的GC含量低,与位于SSC区的NADH基因相关[17]。
SSR又称微卫星,一般是1~6 bp的重复序列,具有高多态性和广泛分布的特点,是高等真核生物叶绿体基因组的重要组成部分,成为研究植物遗传多样性、植物品种鉴定和植物遗传稳定性的重要工具[17]。在本试验中,马家柚叶绿体基因组共检测到79个SSR,包括单核苷酸重复序列和三核苷酸重复序列2种不同类型,其中A和T重复占98.72%,表明马家柚叶绿体基因组SSR偏好使用碱基A和T,这与大多数柑橘属植物叶绿体SSR序列的组成相似。例如,六月早蜜柚叶绿体基因组的SSR为101个,其中68个A和T单碱基重复[21];阿曼酸橙叶绿体基因组的SSR为109个(多为A和T单碱基重复),大多数SSR位于基因间区,少数位于CDS基因(如matK和ycf1)[25]。本研究从马家柚叶绿体基因组中检测到的SSR可说明马家柚的种间多态性,并获知ycf1基因有254个变异位点,变异位点最多。ycf1序列是叶绿体基因组内的片段,由于其进化速率快,序列变异较大,近年来已被陆续应用于兰科[45]等植物的分子鉴定中;且陆地植物物种在PCR成功率、序列位点变异率及物种鉴别率等方面优于matK、rbcL及trnH-psbA等常用叶绿体DNA条形码,因此被认为是最具潜力的陆地植物DNA条形码序列[46]。可为马家柚品种的道地性的分子鉴定提供可靠的遗传标记,为后续对马家柚种质资源的化学成分、抗逆性和其他品质性状进行深入研究,为选育产量高、抗逆性强、品质优的新品种奠定了分子基础。
植物的叶绿体基因组的IR/SC边界时常会发生收缩和扩张,这些收缩和扩张会导致假基因的产生、基因的重复以及基因的缺失[17]。马家柚及其18个同属种叶绿体基因组的变异圈图、mVIST结构变异图和Pi多样性指数分析图显示,LSC区的变异性整体较高,其次是SSC区,IR区变异性最低,10个高变异区域均位于LSC和SSC区,这可能与IR区进化的保守性有关[17]。IR/SC边界的收缩和扩张决定在植物叶绿体基因组的演化进程[17]。徐世荣等[21]的研究表明,芸香科植物的基因结构和基因顺序较为相似,只有JLB(LSC与IRb边界)和JSB(IRb与SSC边界)2个边界存在差异。JLB边界包括2种类型(第1种类型:无基因横跨边界;第2种类型:rpl22基因横跨JLB边界),JSB边界包括3种类型(第1种类型:无基因横跨边界;第2种类型:ycf1基因横跨JSB边界;第3种类型:ndhF基因横跨JSB边界)。Su等[25]的研究表明,阿曼酸橙的IR、LSC和SSC区域之间的连接与血橙(C. sinensis)相似,rpl22基因横跨JLB边界。本试验结果也证实了这种观点,在本试验中,马家柚及其18个同属种叶绿体基因组各个结构极为稳定,未发现明显的IR扩张和收缩,马家柚及其18个同属种叶绿体基因组的JLB边界有rps19基因横跨边界,属于JLB边界的第2种类型;马家柚及其18个同属种叶绿体基因组JSB边界无基因横跨边界,均位于IRb区的ycf1基因和SSC区的ndhF基因之间,属于JSB边界的第1种类型。
植物叶绿体基因组第3位碱基的突变为同义突变。叶绿体基因组密码子的偏好性是指植物使用同义密码子的频率存在差异,这种差异多是由碱基组成所造成的[17]。中性进化理论认为,氨基酸的改变取决于第1位和第2位碱基的非同义突变,不取决于第3位碱基的同义突变,因此,GC3一般作为衡量密码子偏好性的重要指标[17]。本研究中马家柚及其18个同属种叶绿体基因组密码子3个位置GC含量的平均值为39.06%,表明马家柚及其18个同属种叶绿体更倾向于使用A/U密码子,这与徐世荣等[21]对六月早蜜柚叶绿体基因组的研究结果一致。马家柚及其18个同属种叶绿体基因组密码子GC3含量较低,这一特征与六月早蜜柚[21]相同,符合Campbll等[47]提出的假设:高等植物的密码子一般偏好以A/U结尾。ENC表示密码子偏离随机选择的程度,是衡量密码子使用偏好性的重要指标[44]。ENC值越大,密码子使用偏好性越弱,ENC值≤35,密码子偏好性较强[45]。马家柚叶绿体基因组88个CDS基因密码子的ENC值介于28.311~61.000之间,平均值为48.02,84个基因的ENC值>35,4个基因的ENC值<35,密码子偏好性较弱。马家柚叶绿体基因组psbM和petG基因ENC值最高(61),说明这两个基因具有保守的DNA序列,在进化过程中受自然选择影响小;马家柚叶绿体基因组中rpl32基因的ENC值最低(28.311),受突变的影响较小,密码子使用偏好性较强。
植物叶绿体基因组密码子偏好性的影响因子一般有自然选择和内部突变两种。在本试验中,中性绘图分析(GC3-GC12分析)、ENC-plot分析和PR2-plot分析表明在马家柚及其18个同属种在进化过程中,其密码子偏好性主要受自然选择而非受内部突变的影响,究其原因,可能是叶绿体作为进行光合作用的重要细胞器,其基因演化必然要受到自然选择的影响[17]。RSCU分析是一种根据相对密码子偏好性来分析基因表达水平的方法。本研究采用高表达优越密码子方法确定了马家柚叶绿体基因组10个最优密码子,3个以A结尾,7个以U结尾,由此可见马家柚叶绿体基因组中密码子偏好NNA、NNU型。这与徐世荣等[21]对六月早蜜柚叶绿体基因组的研究结果一致,说明柑橘属植物的密码子偏好性存在着一定的相似性,这种密码子使用模式可能是由于柑橘属植物叶绿体基因组密码子的使用偏好性在进化关系上较为保守。一般来说,在正向选择和突变压力较强的情况下,最优密码子的数量较多;而在纯化选择的情况下,最优密码子的数量较少。不同物种的最优密码子及数量不同,说明物种受到的进化压力存在差异[17]。本研究中共确定了10个马家柚叶绿体基因组的最优密码子,数量偏少,因此推测马家柚叶绿体基因组可能处于纯化选择之下。
叶绿体基因组DNA能有效进行物种鉴定和系统亲缘关系分析[17]。徐世荣等[21]和Xu等[22]的研究表明,六月早蜜柚与C. platymamma、柠檬和甜橙聚为一小分支,福建琯溪蜜柚与Low acid pummelo(C. maxima,NC034290.1)聚为一小分支;Zhang等[23]的研究表明,云南红河柑橘与柠檬(C. limon,KY085897.1)、C. platymamma(NC030194.1)、甜橙(C. sinensis,DQ864733.1)和Low acid pummelo(C. maxima,NC034290.1)聚为一分支,但云南红河柑橘单独成一小分支,表明福建琯溪蜜柚与云南红河柑橘存在一定的亲缘关系,但也是一个柑橘属中较为独特的品种;Cai等[24]的研究表明,手指柠檬栽培种与枸橼(C. medica,NC050939.1)聚为一分支,表明两者亲缘关系较近。目前,马家柚的种植区主要集中在江西上饶,现有研究的取样范围和分子标记选择受限,使得马家柚的属内进化关系仍模糊。刘勇[48]通过SSR和AFLP分子鉴定,认为马家柚与广丰周边地区的信木柚亲缘关系较近;徐宸宇等[49]对江西46份柚资源进行了SSR分子鉴定,认为马家柚可能是由广丰本地土柚衍变而成的变异株系。笔者在本试验中,为了进一步揭示马家柚在柑橘属种间的亲缘关系,选取了马家柚及其85个同科种(芸香科柑橘亚科、芸香亚科和飞龙掌血亚科)和3个外群种(橄榄NC048982、苦树MW801117、海人树MK830069),利用FastTree软件GTR模型(Generalized time-reversible model)构建ML系统发育树。结果表明,马家柚(MJY,C. maxima (L.) Osbeck ‘Majiayou)与西双版纳东试早柚(C. maxima,KY055833,来源地:云南)、日本夏橙(C. natsudaidai,ON193075,来源地:韩国)、福建六月早蜜柚(C. maxima ‘Liuyuezao,MT527726,来源地:福建)、福建琯溪蜜柚[C. maxima (Burm.) Merr. ‘Guanximiyou,MN782007,来源地:福建]有亲缘关系,是一个柑橘属中较为独特的品种。
4 结 论
马家柚叶绿体基因组全长160 186 bp,共注释到133个功能基因,共检测到79个SSR和34个Longrepeat。马家柚叶绿体基因组SC/IR边界较为保守,密码子偏好性较弱(主要受自然选择的影响),最优密码子有10个(AAU、UGU、AAA、UUU、GCU、GGA、CCA、ACU、CGU、AGU)。马家柚与西双版纳东试早柚、日本夏橙、福建六月早蜜柚、福建琯溪蜜柚有亲缘关系,是柑橘属中一个较为独特的品种。
参考文献 References:
[1] 林涛,何丽云,王帆,李炳根,阿提克穆·麦麦提,周仁辉,程薪宇. 马家柚花部特征及其访花昆虫种类调查[J]. 环境昆虫学报,2023,45(1):101-113.
LIN Tao,HE Liyun,WANG Fan,LI Binggen,Atikemu·Maimaiti,ZHOU Renhui,CHENG Xinyu. The floral characteristics and its flower-visiting insects species investigation of Citrus maxima (L.) Osbeck cv. ‘Majiayou[J]. Journal of Environmental Entomology,2023,45(1):101-113.
[2] 毛桑隐,路志浩,张祥,叶俊丽,伊华林,柴利军,邓秀新,吴方方,徐强. 花粉直感对马家柚果实品质的影响[J]. 果树学报,2023,40(11):2391-2402.
MAO Sangyin,LU Zhihao,ZHANG Xiang,YE Junli,YI Hualin,CHAI Lijun,DENG Xiuxin,WU Fangfang,XU Qiang. Effect of xenia on fruit quality of Majiayou[J]. Journal of Fruit Science,2023,40(11):2391-2402.
[3] 谢婧蘅,杨莉,旦世浩,邱丽,张王妮,刘德春,胡威,刘勇. 套袋对马家柚果实外观及内在品质的影响[J]. 核农学报,2021,35(1):229-237.
XIE Jingheng,YANG Li,DAN Shihao,QIU Li,ZHANG Wangni,LIU Dechun,HU Wei,LIU Yong. Effect of bagging on fruit appearance and inner quality of Majia pomelo[J]. Journal of Nuclear Agricultural Sciences,2021,35(1):229-237.
[4] 毛小涛,陈凯,孙志锋,樊海敏,张处平. 马家柚光合特性研究[J]. 上饶师范学院学报,2023,43(3):73-78.
MAO Xiaotao,CHEN Kai,SUN Zhifeng,FAN Haimin,ZHANG Chuping. Study on photosynthetic characteristics of Citrus grandis (L.) Osbeck[J]. Journal of Shangrao Normal University,2023,43(3):73-78.
[5] 姜启航,朱凯杰,吴方方,徐娟,徐强,柴利军,邓秀新,叶俊丽. 套袋处理对‘马家柚果实挥发性物质积累的影响[J]. 果树学报,2020,37(11):1701-1710.
JIANG Qihang,ZHU Kaijie,WU Fangfang,XU Juan,XU Qiang,CHAI Lijun,DENG Xiuxin,YE Junli. Effects of fruit bagging on the accumulation of volatile compounds in ‘Majiayou pumelo[J]. Journal of Fruit Science,2020,37(11):1701-1710.
[6] 邱丽,杨莉,旦世浩,刘德春,胡威,张王妮,刘勇. 套袋对‘马家柚果肉主要类胡萝卜素积累及相关基因表达的影响[J]. 果树学报,2020,37(2):153-163.
QIU Li,YANG Li,DAN Shihao,LIU Dechun,HU Wei,ZHANG Wangni,LIU Yong. Effects of bagging on the accumulation of main carotenoids and the related gene expression in ‘Majiayou pomelo[J]. Journal of Fruit Science,2020,37(2):153-163.
[7] 易明亮,张王妮,杨莉,匡柳青,刘德春,刘勇,胡威. ‘马家柚果实发育期有机酸含量变化及柠檬酸代谢相关基因的表达分析[J]. 江西农业大学学报,2022,44(4):841-851.
YI Mingliang,ZHANG Wangni,YANG Li,KUANG Liuqing,LIU Dechun,LIU Yong,HU Wei. Analysis of organic acid content and expression of citric acid metabolism related genes during fruit development of ‘Majia pomelo[J]. Acta agriculturae universitatis Jiangxiensis,2022,44(4):841-851.
[8] 周丽明,韩金多. 马家柚果酒发酵工艺及其抗氧化作用分析[J]. 南方农业学报,2018,49(2):348-353.
ZHOU Liming,HAN Jinduo. Fermentation process and antioxidant effects of Majia pummelo wine[J]. Journal of Southern Agriculture,2018,49(2):348-353.
[9] 杨莉,张涓涓,刘德春,刘山蓓,徐炳星,周施清,毛卫平,刘勇. 马家柚粗皮果形成过程的解剖学观察[J]. 经济林研究,2017,35(3):152-155.
YANG Li,ZHANG Juanjuan,LIU Dechun,LIU Shanbei,XU Bingxing,ZHOU Shiqing,MAO Weiping,LIU Yong. Anatomically observation on forming process of rough fruits in Citrus grandis[J]. Nonwood Forest Research,2017,35(3):152-155.
[10] 张涓涓,杨莉,刘德春,刘山蓓,徐炳星,周施清,毛卫平,刘勇. 土壤养分状况与马家柚果实品质相关性的多元分析[J]. 经济林研究,2015,33(4):25-31.
ZHANG Juanjuan,YANG Li,LIU Dechun,LIU Shanbei,XU Bingxing,ZHOU Shiqing,MAO Weiping,LIU Yong. Multivariate analysis of relationship between soil nutrient status and fruit quality of Majia shaddock[J]. Nonwood Forest Research,2015,33(4):25-31.
[11] SLOAN D B,TRIANT D A,FORRESTER N J,BERGNER L M,WU M,TAYLOR D R. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae)[J]. Molecular Phylogenetics and Evolution,2014,72:82-89.
[12] NAZARENO A G,CARLSEN M,LOHMANN L G. Complete chloroplast genome of Tanaecium tetragonolobum:The first Bignoniaceae plastome[J]. PLoS One,2015,10(6):e0129930.
[13] LI D M,LI J,WANG D R,XU Y C,ZHU G F. Molecular evolution of chloroplast genomes in subfamily Zingiberoideae (Zingiberaceae)[J]. BMC Plant Biology,2021,21(1):558.
[14] LIANG H,ZHANG Y,DENG J B,GAO G,DING C B,ZHANG L,YANG R W. The complete chloroplast genome sequences of 14 Curcuma species:Insights into genome evolution and phylogenetic relationships within Zingiberales[J]. Frontiers in Genetics,2020,11:802.
[15] KATHRIARACHCHI H,HOFFMANN P,SAMUEL R,WURDACK K J,CHASE M W. Molecular phylogenetics of Phyllanthaceae inferred from five genes (plastid atpB,matK,3'ndhF,rbcL,and nuclear PHYC))[J]. Molecular Phylogenetics and Evolution,2005,36(1):112-134.
[16] DANIELL H,LIN C S,YU M,CHANG W J. Chloroplast genomes:Diversity,evolution,and applications in genetic engineering[J]. Genome Biology,2016,17(1):134.
[17] 朱强龙,朱子成,王鹏飞,吕慧玲,崔浩楠,栾非时. 葫芦科作物线粒体和叶绿体基因组研究进展[J]. 中国瓜菜,2016,29(8):1-8.
ZHU Qianglong,ZHU Zicheng,WANG Pengfei,L? Huiling,CUI Haonan,LUAN Feishi. Advances on mitochondria and chloroplast genomes of cucurbit crop[J]. China Cucurbits and Vegetables,2016,29(8):1-8.
[18] ABDULLAH,MEHMOOD F,RAHIM A,HEIDARI P,AHMED I,POCZAI P. Comparative plastome analysis of Blumea,with implications for genome evolution and phylogeny of Asteroideae[J]. Ecology and Evolution,2021,11(12):7810-7826.
[19] LIU H,HUANG Y,DU X,CHEN Z,ZENG X,CHEN Y,ZHANG H. Patterns of synonymous codon usage bias in the model grass Brachypodium distachyon[J]. Genetics and Molecular Research,2012,11(4):4695-4706.
[20] QI Y Y,XU W J,XING T,ZHAO M M,LI N N,YAN L,XIA G M,WANG M C. Synonymous Codon usage bias in the plastid genome is unrelated to gene structure and shows evolutionary heterogeneity[J]. Evolutionary Bioinformatics Online,2015,11:65-77.
[21] 徐世荣,陈燕琼,潘东明,潘鹤立. 六月早蜜柚叶绿体基因组及其特征分析[J]. 热带作物学报,2021,42(5):1223-1230.
XU Shirong,CHEN Yanqiong,PAN Dongming,PAN Heli. Chloroplast genome sequence and characteristics analysis of Citrus maxima ‘Liuyuezao[J]. Chinese Journal of Tropical Crops,2021,42(5):1223-1230.
[22] XU S R,HUANG C Y,DENG Y T,ZHOU L L,PAN D M,PAN H L. The complete chloroplast genome sequence of Citrus maxima (Burm.) Merr. ‘Guanximiyou[J]. Mitochondrial DNA. Part B,Resources,2020,5(1):482-483.
[23] ZHANG Z H,LONG C R,JIANG Y,BEI X J,WANG S H. Characterization of the complete chloroplast genome of Citrus hongheensis,a key protected wild plant in Yunnan Province of China[J]. Mitochondrial DNA. Part B,Resources,2020,5(3):3514-3515.
[24] CAI Q N,WANG H X,CHEN D J,KE X R,ZHU Z X,WANG H F. The complete chloroplast genome sequence of a Citrus australasica cultivar (Rutaceae)[J]. Mitochondrial DNA. Part B,Resources,2021,7(1):54-55.
[25] SU H J,HOGENHOUT S A,AL-SADI A M,KUO C H. Complete chloroplast genome sequence of Omani lime (Citrus aurantiifolia) and comparative analysis within the rosids[J]. PLoS One,2014,9(11):e113049.
[26] ISHIKAWA R,BADENOCH N,MIYAGI K,MEDORUMA K,OSADA T,ONISHI M. Multi-lineages of Shiikuwasha (Citrus depressa Hayata) evaluated by using whole chloroplast genome sequences and its bio-diversity in Okinawa,Japan[J]. Breeding Science,2016,66(4):490-498.
[27] BAUSHER M G,SINGH N D,LEE S B,JANSEN R K,DANIELL H. The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck ‘Ridge Pineapple:Organization and phylogenetic relationships to other angiosperms[J]. BMC Plant Biology,2006,6:21.
[28] DOYLE J J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue[J]. Phytochemical Bulletin,1987,19(1):11-15.
[29] CHEN S F,ZHOU Y Q,CHEN Y R,GU J. Fastp:An ultra-fast all-in-one FASTQ preprocessor[J]. Bioinformatics,2018,34(17):i884-i890.
[30] DIERCKXSENS N,MARDULYN P,SMITS G. NOVOPlasty:De novo assembly of organelle genomes from whole genome data[J]. Nucleic Acids Research,2017,45(4):e18.
[31] HUANG X,MADAN A. CAP3:A DNA sequence assembly program[J]. Genome Research,1999,9(9):868-877.
[32] TILLICH M,LEHWARK P,PELLIZZER T,ULBRICHT-JONES E S,FISCHER A,BOCK R,GREINER S. GeSeq-versatile and accurate annotation of organelle genomes[J]. Nucleic Acids Research,2017,45(W1):W6-W11.
[33] LOWE T M,EDDY S R. tRNAscan-SE:A program for improved detection of transfer RNA genes in genomic sequence[J]. Nucleic Acids Research,1997,25(5):955-964.
[34] LOHSE M,DRECHSEL O,BOCK R. OrganellarGenomeDRAW (OGDRAW):A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes[J]. Current Genetics,2007,52(5/6):267-274.
[35] GRANT J R,STOTHARD P. The CGView Server:A comparative genomics tool for circular genomes[J]. Nucleic Acids Research,2008,36(Web Server issue):W181-W184.
[36] THIEL T,MICHALEK W,VARSHNEY R,GRANER A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.)[J]. Theoretical and Applied Genetics,2003,106(3):411-422.
[37] KURTZ S,CHOUDHURI J V,OHLEBUSCH E,SCHLEIERMACHER C,STOYE J,GIEGERICH R. REPuter:The manifold applications of repeat analysis on a genomic scale[J]. Nucleic Acids Research,2001,29(22):4633-4642.
[38] SHARP P M,LI W H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘Rare codons[J]. Nucleic Acids Research,1986,14(19):7737-7749.
[39] PETKAU A,STUART-EDWARDS M,STOTHARD P,VAN DOMSELAAR G. Interactive microbial genome visualization with GView[J]. Bioinformatics,2010,26(24):3125-3126.
[40] AMIRYOUSEFI A,HYV?NEN J,POCZAI P. IRscope:An online program to visualize the junction sites of chloroplast genomes[J]. Bioinformatics,2018,34(17):3030-3031.
[41] ROZAS J,ROZAS R. DnaSP,DNA sequence polymorphism:An interactive program for estimating population genetics parameters from DNA sequence data[J]. Bioinformatics,1995,11(6):621-625.
[42] KATOH K,STANDLEY D M. MAFFT multiple sequence alignment software version 7:Improvements in performance and usability[J]. Molecular Biology and Evolution,2013,30(4):772-780.
[43] PRICE M N,DEHAL P S,ARKIN A P. FastTree 2:Approximately maximum-likelihood trees for large alignments[J]. PLoS One,2010,5(3):e9490.
[44] 伍泓昆. 基于DArT芯片及叶绿体基因组分析的柑橘属植物进化与分类研究[D]. 重庆:西南大学,2016.
WU Hongkun. Evaluation and phylogenetic relationship of Citrus L. based on DArT markers and chloroplast genomes analyses[D]. Chongqing:Southwest University,2016.
[45] NEUBIG K M,WHITTEN W M,CARLSWARD B S,BLANCO M A,ENDARA L,WILLIAMS N H,MOORE M. Phylogenetic utility of ycf1 in orchids:A plastid gene more variable than matK[J]. Plant Systematics and Evolution,2009,277(1/2):75-84.
[46] DONG W P,XU C,LI C H,SUN J H,ZUO Y J,SHI S,CHENG T,GUO J J,ZHOU S L. ycf1,the most promising plastid DNA barcode of land plants[J]. Scientific Reports,2015,5:8348.
[47] CAMPBELL W H,GOWRI G. Codon usage in higher plants,green algae,and cyanobacteria[J]. Plant Physiology,1990,92(1):1-11.
[48] 刘勇. 柚类资源分子系统学及其核心种质构建研究[D]. 武汉:华中农业大学,2005.
LIU Yong. Molecular phylogenetic analysis and core collection construction using SSR and AFLP markers in pummelo[D]. Wuhan:Huazhong Agricultural University,2005.
[49] 徐宸宇,曹立新,唐启正,吴巨勋,伊华林. 马家柚遗传来源鉴定与适宜授粉品种筛选[J]. 华中农业大学学报(自然科学版),2022,41(2):124-135.
XU Chenyu,CAO Lixin,TANG Qizheng,WU Juxun,YI Hualin. Identification of Majia pomelo germplasm and screening of varieties with suitable pollination[J]. Journal of Huazhong Agricultural University (Natural Science Edition),2022,41(2):124-135.
