超高效液相色谱-串联质谱法测定调节三高类保健食品中59 种非法添加药物
2024-05-20邵瑞婷丁学妍
邵瑞婷,丁学妍,姜 洁
(北京市食品检验研究院(北京市食品安全监控和风险评估中心),国家市场监管重点实验室(食品安全重大综合保障关键技术),北京 100041)
“三高症”指高血压、高血糖和高脂血症。现在已经成为威胁人们身体健康的常见疾病[1]。“三高”症状既可能单独存在,又常相互关联,只要出现其中任何一种,后期都易形成“三高症”[2-3]。为了降低患病概率,声称具有辅助调节“三高”功效的保健食品受到人们的追捧。不法分子为增强“疗效”,向其中添加药物,这些药物的摄入可能会对人们的身体健康造成不可逆的危害[4-5]。
近几年,原国家食品药品监督管理总局连续颁布了补充检验方法和批准件,针对“三高”的6 个官方标准中收载了28 种常见的降血压、降血糖以及降血脂药物[6-11],鉴于治疗“三高症”的药物使用品种增多,部分熟悉检测标准、通晓药理药效的不法分子为了逃避监管,存在添加非规定检测标准涵盖药物的情况,同时鉴于补充检验方法中,药物覆盖面欠缺的问题,坚持以问题为导向,有必要根据药物的疗效,扩充“三高症”治疗的常见化学药物的高通量检测方法,最大可能性避免漏检,一定程度上扩大对违法添加化学药品的打击面[12-13]。
本研究将上述原国家食品药品监督管理总局连续颁布的补充检验方法和批准件的化学药品纳入同一方法进行研究,并另选取文献报道有检出或者根据药品疗效及可获得性、添加可能性较大的化学药品总计59 种进行同时测定研究。包括降高血压药物:布美他尼、吲达帕胺、妥拉唑林、酚妥拉明、育亨宾、哌唑嗪、特拉唑嗪、卡托普利、可乐定、阿替洛尔、硝苯地平、尼群地平、非洛地平、尼索地平、氨氯地平、尼莫地平、利血平、普萘洛尔、美托洛尔、卡拉洛尔、倍他洛尔、纳多洛尔、卡维地洛、依普沙坦、厄贝沙坦、颉沙坦、坎替沙坦、莫西普利、替米沙坦、氯噻嗪、氢氯噻嗪、呋塞米、普伐他汀[14-15];降血糖类药物:二甲双胍、丁二胍、苯乙双胍、苯扎贝特、甲苯磺丁脲、格列齐特、吡格列酮、罗格列酮、格列波脲、瑞格列奈、格列美脲、格列苯脲、格列喹酮、格列吡嗪、维达他汀、西他列汀、氯磺丙脲[16-17];降血脂类药物:烟酸、非诺贝特、洛伐他汀、辛伐他汀、脱羟基洛伐他汀、美伐他汀、洛伐他汀羟酸钠盐、阿伐他汀钙盐、西立伐他汀钠盐[18-19]。前期通过检索文献可知,针对食品中违法添加的检测方法主要有拉曼法[20]、毛细管电泳法[21]、荧光光谱法[22]、近红外光谱法[23]、气相色谱-质谱法[24]、气相色谱-串联质谱法[25]、高效薄层色谱法[26]、高效液相色谱法[27-28]和液相色谱-串联质谱法[29-30]等。基于液相色谱-质谱联用法较好的定性、定量能力,其在多组分农残和兽残检测中已得到广泛应用,建立了多项食品检测国家标准[31-32]。而其在保健食品非法添加检测上未有相关国家标准支持,为便于开展多组分非法添加药物检测,本研究旨在建立一个高效、快速的液相色谱-质谱联用方法,以期实现59 种违法添加“三高”类药物的同时检测。
1 材料与方法
1.1 材料与试剂
5982-4921 QuEChERS(quick,easy,cheap,effective,rugged and safe)管、5982-0028 QuEChERS管、5982-5122 QuEChERS管、Agilent 0.2 μm微孔滤膜(聚四氟乙烯(polytetrafluoroethylene,PTFE))、0.2 μm微孔滤膜(尼龙)美国Agilent公司;Titan3 0.2 μm微孔滤膜(PTFE)、Titan3 0.2 μm微孔滤膜(尼龙)美国Thermo Fisher Scientific公司;聚丙烯(GH polypro,GHP)0.2 μm微孔滤膜 美国Waters公司;Agela 0.22 μm微孔滤膜(尼龙)天津Agela公司;xiboshi 0.20 μm微孔滤膜(尼龙)天津市希波氏科技有限公司;Jinteng 0.22 μm微孔滤膜(尼龙)天津市津腾实验设备有限公司。
乙腈、甲醇、乙酸铵(均为色谱纯)美国Thermo Fisher Scientific公司;甲酸(质谱级)上海阿拉丁生化科技股份有限公司;烟酸、非诺贝特、洛伐他汀、辛伐他汀、脱羟基洛伐他汀、美伐他汀、阿伐他汀钙盐、西立伐他汀钠盐、二甲双胍、丁二胍、苯扎贝特、格列齐特、吡格列酮、维达他汀、西他列汀、氯磺丙脲、布美他尼、吲达帕胺、妥拉唑林、酚妥拉明、哌唑嗪、卡托普利、可乐定、阿替洛尔、硝苯地平、尼群地平、非洛地平、尼索地平、氨氯地平、尼莫地平、普萘洛尔、美托洛尔、卡拉洛尔、倍他洛尔、纳多洛尔、卡维地洛、依普沙坦、厄贝沙坦、颉沙坦、坎替沙坦、莫西普利、替米沙坦、氯噻嗪、氢氯噻嗪、呋塞米、普伐他汀(100 μg/mL)、洛伐他汀羟酸钠盐(纯度98%)、苯乙双胍(纯度98%)、甲苯磺丁脲(纯度99.9%)、罗格列酮(纯度98%)、格列波脲(纯度99.1%)、瑞格列奈(纯度99.9%)、格列美脲(纯度99.7%)、格列喹酮(纯度98%)、格列吡嗪(纯度98%)、利血平(纯度99.4%)天津阿尔塔科技有限公司;格列苯脲(纯度99%)、盐酸育亨宾(纯度99.4%)标准品 德国Dr.Ehrenstorfer GmbH公司;特拉唑嗪(纯度98%)标准品 德国Sigma-Aldrich公司;合辉安泰片(葡萄味)东营大振生物工程有限公司;口服液 上海大正力保健有限公司;茶包 北京同仁堂兴安保健科技有限责任公司。
1.2 仪器与设备
ACQUITYTMTQ-XS超高效液相色谱-串联质谱仪美国Waters公司;均质器 德国IKA公司;高速冷冻离心机 美国Thermo Fisher Scientific公司;超声波提取仪 中国五洲东方公司;万分之一电子天平 瑞士梅特勒-托利多公司。
1.3 方法
1.3.1 标准品配制
精确称取各类固体标准品适量、精密量取各类液体标准品适量,分别置于10 mL棕色容量瓶中,用甲醇溶解、混匀并定容,配制成1 mg/mL的标准储备溶液,避光、-18 ℃保存。
精密吸取各类外标标准储备溶液,用甲醇定容至10 mL,配制成混合标准溶液,其中非诺贝特、洛伐他汀、辛伐他汀、苯扎贝特、布美他尼、吲达帕胺、酚妥拉明、育亨宾、哌唑嗪、特拉唑嗪的质量浓度为1 μg/mL;烟酸、脱羟基洛伐他汀、美伐他汀、阿伐他汀钙盐、西立伐他汀钠盐、二甲双胍、丁二胍、苯乙双胍、甲苯磺丁脲、格列齐特、吡格列酮、罗格列酮、格列波脲、瑞格列奈、格列美脲、格列苯脲、格列喹酮、格列吡嗪、维达他汀、西他列汀、卡托普利、可乐定、阿替洛尔、硝苯地平、尼群地平、非洛地平、尼索地平、氨氯地平、尼莫地平、利血平、普萘洛尔、美托洛尔、卡拉洛尔、倍他洛尔、纳多洛尔、卡维地洛、依普沙坦、厄贝沙坦、颉沙坦、坎替沙坦、莫西普利、替米沙坦的质量浓度为2 μg/mL;氯噻嗪、氢氯噻嗪、呋塞米、氯磺丙脲、普伐他汀的质量浓度为5 μg/mL;妥拉唑林的质量浓度为4 μg/mL;脱羟基洛伐他汀的质量浓度为50 μg/mL。避光、-18 ℃保存。
准确吸取混合标准溶液1 mL用甲醇定容至10 mL,配制成混合标准中间液,现用现配。
吸取混合标准溶液2.5 mL,于50 mL容量瓶中,甲醇定容至刻度线,配制成混合标准使用液,混匀,备用。
1.3.2 样品前处理
称取0.5 g试样(精确0.01 g)置于10 mL具塞玻璃比色管中,加入8 mL甲醇,涡旋振荡,超声提取20 min,冷却至室温后,用甲醇定容至刻度线,摇匀,取5 mL提取液于10 mL塑料离心管中,以10 000 r/min离心10 min,取1.5 mL上清液过QuEChERS管净化,收集1 mL净化液,氮吹至近干,用1 mL 40%甲醇溶液复溶,过0.2 μm PTFE滤膜后进行仪器检测。
基质匹配混合标准系列溶液:称取8 份0.5 g(精确0.01 g)的同一试样,分别加入混合标准中间液0、0.025、0.05、0.1、0.2、0.5、0.8、1 mL,采用与试样相同的前处理方法,得到基质匹配混合标准系列溶液,备用。
1.3.3 液相色谱-串联质谱条件
1.3.3.1 色谱条件
色谱柱:ACQUITY HSS T3(100 mm×2.1 mm,1.8 μm);柱温:30 ℃;进样体积:10 μL;流速:0.3 mL/min;流动相:A相为乙腈、B相为0.1%甲酸溶液(5 mmol/L乙酸铵溶液);梯度洗脱程序:0~1 min,5% A、95% B;1~2 min,5%~30% A、95%~70% B;2~5 min,30%~90% A、70%~10% B;5~6.5 min,90% A、1 0% B;6.5~8 min,9 0%~5% A、10%~95% B;8~10 min,5% A、95% B。
1.3.3.2 质谱条件
离子源:电喷雾电离源(electrospray ionization,ESI);毛细管电压:3.0 kV;锥孔电压:20 V;离子源温度:150 ℃;脱溶剂气温度:350 ℃;脱溶剂气流速:650 L/h;锥孔气流速:150 L/h;碰撞气流速:0.15 mL/min。其他质谱条件参数见表1。

表1 59 种非法添加药物质谱条件参数Table 1 Mass spectrometric parameters for detection of 59 illegally added drugs
1.3.4 条件优化
1.3.4.1 定容液的选择
本研究以体积分数5%、10%、20%、30%、40%、50%、60%、70%、80%、90%甲醇溶液以及甲醇作为定容液,考察定容液对目标物的影响。分别吸取定容液0.95 mL置于2 mL玻璃瓶中,加入混合标准中间液0.05 mL,涡旋振荡后供仪器检测。
1.3.4.2 超声时间优化
称取5 个0.5 g阴性片剂样品置于10 mL玻璃具塞比色管中,涡旋振荡,分别加入混合标准中间液0.2 mL,加入8 mL甲醇溶液,涡旋混匀后,分别超声提取10、20、30、40、50 min,冷却至室温后,定容至10 mL,取1 mL提取液氮吹至近干,用1 mL 40%甲醇溶液复溶后,以12 000 r/min离心5 min,取上清液供仪器检测,用以考察超声时间对目标物提取效果的影响。
1.3.4.3 基质效应(matrix effect,ME)评估
采用下式对59 种化合物的ME进行评价[33]:
1.3.4.4 质谱条件优化
考虑到离子扫描模式对目标物响应的影响,将混合标准使用液不接色谱柱直接注射,在正离子(ESI+)和负离子(ESI-)模式下进行全扫描以选择适当的电离方式。经实验考察,本研究选取正、负离子模式,并结合多反应监测模式采集目标化合物的离子信息。
1.4 数据处理与分析
使用TQ-XS MassLynx软件进行质谱分析、数据采集和处理,采用Excel 2010软件进行数据统计和图表绘制。
2 结果与分析
2.1 定容液的选择
本研究对比5%、10%、20%、30%、40%、50%、60%、70%、80%、90%甲醇溶液以及甲醇作为定容液分离脱羟基洛伐他汀、氯噻嗪的效果,结果见图1,以40%甲醇溶液作为定容液时,59 种非法添加药物能够更好地分离,峰型尖锐,且响应值更高,其中,脱羟基洛伐他汀在低于40%甲醇溶液作为定容液时响应值及峰型差异性显著,高于40%甲醇溶液后无明显差异;氯噻嗪在高于40%甲醇溶液后,随着有机相比例升高峰型变差,而低于40%甲醇溶液时无明显变化。故本研究中,选取40%甲醇溶液作为定容液。

图1 不同定容液条件下脱羟基洛伐他汀(a)、氯噻嗪(b)离子色谱图Fig.1 Ion chromatograms of dihydroxylovastatin (a) and chlorthiazide (b) at different final solution volumes
2.2 微孔滤膜的选择
分别吸取1 mL混合标准使用液通过Titan3 PTFE、Titan3尼龙、GHP、Agela尼龙、Agilent PTFE、Agilent尼龙、xiboshi尼龙、Jinteng尼龙滤膜后进行仪器检测,通过8 种微孔滤膜后59 种非法添加药物的回收率分别在<60%、60%~120%、>120% 3 种范围的数量占比如图2所示,采用Titan3 PTFE滤膜各物质回收率在60%~120%范围的占比最高。故在本研究中,选取Titan3 PTFE滤膜作为微孔滤膜。
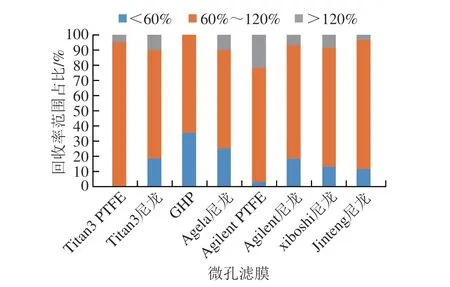
图2 不同微孔滤膜对59 种非法添加药物的回收率范围占比Fig.2 Recoveries of 59 illegally added drugs with different microporous membranes
2.3 超声时间的选择
如图3所示,各组分在20 min的提取回收率相对较高,继续延长超声时间,无法进一步显著提升目标物回收率,反而大多数目标物的回收率会持平或降低,故本研究超声时间选择20 min。

图3 不同超声时间条件下59 种非法添加药物的回收率Fig.3 Recoveries of 59 illegally added drugs under different sonication time conditions
2.4 净化柱的选择
ME是指样品中除了目标分析物以外的其他成分对待测物测定值的影响。本研究主要针对口服液、片剂、茶包等成分复杂的保健食品,样品可能存在较强的ME。一般而言,当ME在80%~120%之间时,ME不明显,在实际检测中可忽略不计;反之则应考虑ME的影响。以口服液样品为例,59 种非法添加药物在样品中的ME分布情况如图4a所示,多种化合物检测受到ME的影响较大。为降低ME的影响,本研究通过对样品提取液进行净化,选取便于批量操作、成本较低且普适性强的QuEChERS净化法[34],比较了5982-4921、5982-0028、5982-5122 3 种成品QuEChERS管对59 种非法添加药物的净化效果,结果见图5。5982-0028管、5982-5122管对洛伐他汀、辛伐他汀、美伐他汀、洛伐他汀羟酸钠盐等净化效果均不理想;5982-4921管对59 种非法添加药物净化回收率均在70%~147.5%。故本实验选用5982-4921 QuEChERS管对提取液进行净化处理,净化后ME分布情况如图4b所示,ME有明显改善,但依旧明显,为了能更准确地测定目标化合物,采用基质匹配标准曲线进行定量分析,进一步降低ME,如图4c所示,ME得到明显改善。
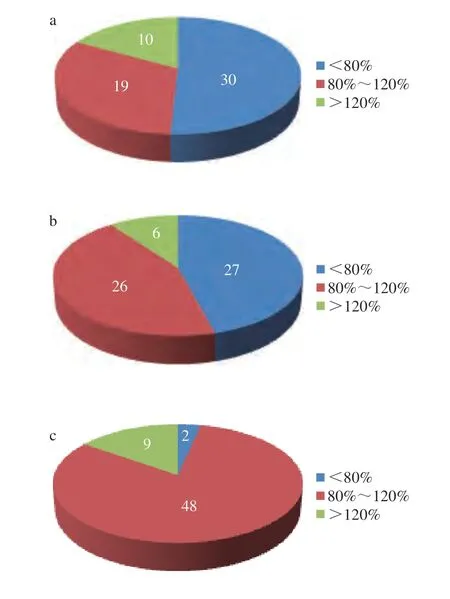
图4 不同处理后口服液样品中59 种非法添加药物的MEFig.4 ME of 59 illegally added drugs in oral liquid samples after different treatments

图5 不同净化柱对59 种非法添加药物回收率的影响Fig.5 Effect of clean-up columns on recoveries of 59 illegally added drugs
2.5 色谱条件的优化
2.5.1 质谱条件优化
如表1所示,氯磺丙脲、氯噻嗪、氢氯噻嗪、呋塞米、普伐他汀在负离子模式下离子化效率较高,灵敏度较好;其他54 种化合物均在正离子模式下,离子化效率较高,灵敏度较好。
2.5.2 色谱条件优化
实验比较了ACQUITY HSS T3(100 mm×2.1 mm,1.8 μm)色谱柱和ACQUITY BEH C18(100 mm×2.1 mm,1.7 μm)色谱柱在相同参数条件下以特拉唑嗪、苯乙双胍、烟酸为例的离子色谱图,如图6所示,BEH C18色谱柱拖尾现象明显,且无法实现多种化合物的基线分离;而HSS T3色谱柱的峰形尖锐,且分离效果明显优于BEH C18色谱柱。故在本实验中选择HSS T3色谱柱。
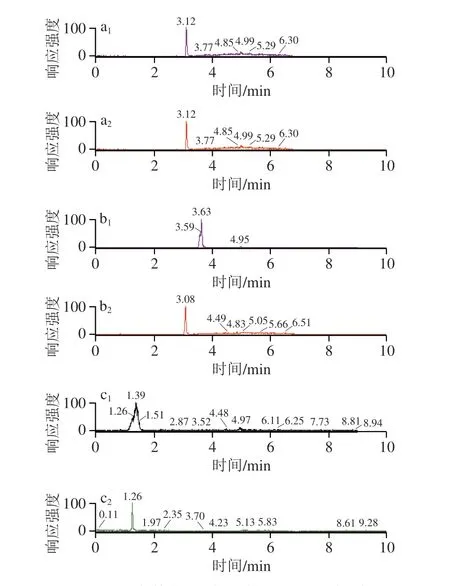
图6 不同色谱柱条件下特拉唑嗪、苯乙双胍、烟酸的离子色谱图Fig.6 Ion chromatograms of 59 illegally added drugs on different columns
2.6 方法学实验
2.6.1 检出限、定量限与线性范围
以待测化合物定性离子对的重构离子色谱峰的信噪比大于或等于3确定方法检出限,以定量离子对的重构离子色谱峰的信噪比大于或等于10确定方法定量限。如表2所示,59 种非法添加药物在线性范围内关系良好,决定系数(R2)均大于0.980,能满足定量分析的要求。对于非诺贝特、洛伐他汀、辛伐他汀、苯扎贝特、布美他尼、吲达帕胺、酚妥拉明、育亨宾、哌唑嗪、特拉唑嗪线性范围为0.25~10 ng/mL,检出限(limit of detection,LOD)为5 μg/kg,定量限(limit of quantitation,LOQ)为10 μg/kg;烟酸、脱羟基洛伐他汀、美伐他汀、阿伐他汀钙盐、西立伐他汀钠盐、二甲双胍、丁二胍、苯乙双胍、甲苯磺丁脲、格列齐特、吡格列酮、罗格列酮、格列波脲、瑞格列奈、格列美脲、格列苯脲、格列喹酮、格列吡嗪、维达他汀、西他列汀、卡托普利、可乐定、阿替洛尔、硝苯地平、尼群地平、非洛地平、尼索地平、氨氯地平、尼莫地平、利血平、普萘洛尔、美托洛尔、卡拉洛尔、倍他洛尔、纳多洛尔、卡维地洛、依普沙坦、厄贝沙坦、颉沙坦、坎替沙坦、莫西普利、替米沙坦线性范围为0.5~20 ng/mL,LOD为10 μg/kg,LOQ为20 μg/kg;氯噻嗪、氢氯噻嗪、呋塞米、氯磺丙脲、普伐他汀线性范围为1.25~50 ng/mL,LOD为25 μg/kg,LOQ为50 μg/kg;妥拉唑林检线性范围为1~40 ng/mL,LOD为20 μg/kg,LOQ为40 μg/kg;洛伐他汀羟酸钠盐线性范围为12.5~500 ng/mL,LOD为250 μg/kg,LOQ为500 μg/kg。
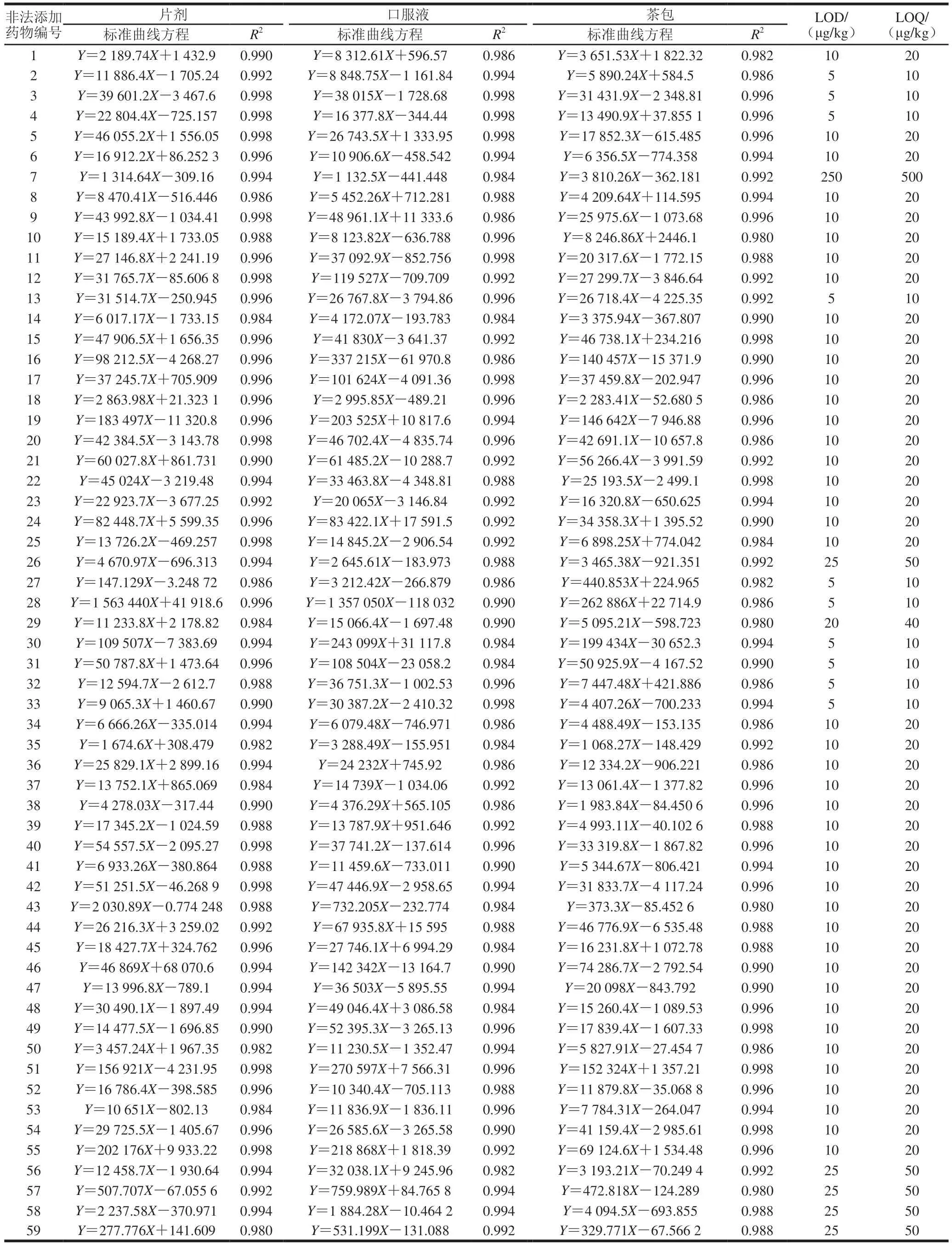
表2 59 种非法添加药物线性参数、LOD与LOQTable 2 Calibration curve equations,LOD and LOQ of 59 illegally added drugs
2.6.2 方法回收率及精密度
分别称取片剂、口服液、茶包阴性基质样品,按照定量限、2 倍定量限、5 倍定量限进行3水平添加试验(n=8),计算回收率及相对标准偏差(relative standard deviation,RSD),如表3所示,本方法所测得样品的加标回收率在60.2%~119.5%之间,RSD在1.2%~15.0%之间。
2.7 实际样品检测
随机抽取市售的调节三高类保健食品5 份,应用本方法进行测定,检测结果均为阴性。阴性样品离子色谱见图7。

图7 阴性样品离子色谱图Fig.7 Ion chromatogram of negative samples
3 结论
本研究选用超高效液相色谱-串联质谱联用技术,针对调节三高类保健食品中可能出现的非法添加成分,建立了同时测定59 种非法添加药物的方法。通过优化色谱条件和质谱参数得到最优的检测条件;优化样品定容液、微孔滤膜、提取时间及净化管对目标物实现了较好的提取和富集。该方法样品前处理操作简单、灵敏度高、准确度高、重现性好,可为调节三高类保健食品的质量安全监管提供技术支持。
