First identification of a novel Aichivirus D in goats with diarrhea
2024-05-13ChenYangKehamoAbiHuaYueFalongYangChengTang
Chen Yang ,Keha-mo Abi ,Hua Yue ,Falong Yang ,Cheng Tang#
1 College of Animal & Veterinary Sciences,Southwest Minzu University,Chengdu 610041,China
2 Key Laboratory of Qinghai-Tibetan Plateau Animal Genetic Resource Reservation and Utilization,Chengdu 610041,China
Kobuvirus comprises 6 officially recognized species,namely Aichivirus A-F,and can be further divided into 20 genotypes through VP1 gene phylogenetic analysis (https://ictv.global/report/chapter/picornaviridae/picornaviridae/kobuvirus).Aichivirus A in human,Aichivirus B in bovine,Aichivirus C in porcine and caprine,Aichivirus D in yak have been proved to be associated with diarrhea (Chen Y Set al.2013;Yanget al.2015;Zhuet al.2016;Zhaiet al.2017;Wanget al.2020;Abiet al.2022;Yanet al.2023).
To date,two Kobuvirus species have been identified in sheep: Aichivirus B and D.Aichivirus B has been found in fecal samples from both healthy and diarrheal sheep (Reuteret al.2010;Barryet al.2011),and Aichivirus D has been detected in Tibetan sheep on the Qinghai-Tibet Plateau,China,by our research team (Abiet al.2021).However,only one kobuvirus species (Aichivirus C) has been found in goats in 4 countries,including South Korean,Italy,China and the USA (Oemet al.2014;Melegariet al.2016;Abiet al.2020;Sobhyet al.2020).
Aichivirus D comprises two officially recognized genotypes (D1 and D2) and two potential novel genotypes (D3 and D4) according to genomic phylogenetic analysis.Aichivirus D1 and D2 have been detected in bovines (Otomaruet al.2016),while the potential Aichivirus D3 has been identified in sheep (Abiet al.2021),and the potential Aichivirus D4,which has been proved to be a diarrhea-causing virus,was detected in yaks (Yanet al.2023).
In June 2021,a goat farm in Sichuan Province,China,experienced a diarrhea outbreak.To investigate possible viruses associated with goat diarrhea,10 goats (aged 2 d to 2 wk) diarrheal samples were collected and pooled for metagenomic sequencing.The results showed that 7 contigs were identified as kobuvirus sequences and shared the highest similarity to ovine Aichivirus D SWUN/AB18/2019 (GenBank accession number MW296158).Based on these contigs,12 overlapping PCR primer pairs (Appendix A) were designed,and a nearly full genome of strain SWUN/B12/2022 (GenBank accession number: OP376931) was obtained from a goat diarrheal sample.The genome sequence is 8,461 nt in length with a 792 nt 5´UTR and a 145 nt 3´UTR,and contains a 7,524 bp complete ORF encoding a polyprotein consisting of structural protein P1 (VP0,VP3,and VP1),nonstructural proteins P2 (2A-2C) and P3 (3A-3D).A phylogenetic tree based on the genome sequence revealed that strain SWUN/B12/2022 is most closely related to ovine Aichivirus D SWUN/AB18/2019 (Fig.1-A).The homology between the two strains were 87.4% in nt sequence and 90.4% in the complete polyprotein: 85.6% in P1,94.1% in 2C,and 92.8% in 3CD protein sequences,respectively (Appendix B).Therefore,SWUN/B12/2022 should be classified into Aichivirus species D according to the ICTV species classification criteria of kobuvirus (Kang and Kim 2018).This is the first report of Aichivirus D detection in goats and obtaining the genome sequence of goat Aichivirus D,contributing to better understanding of the host range and genetic evolution of Aichivirus D.
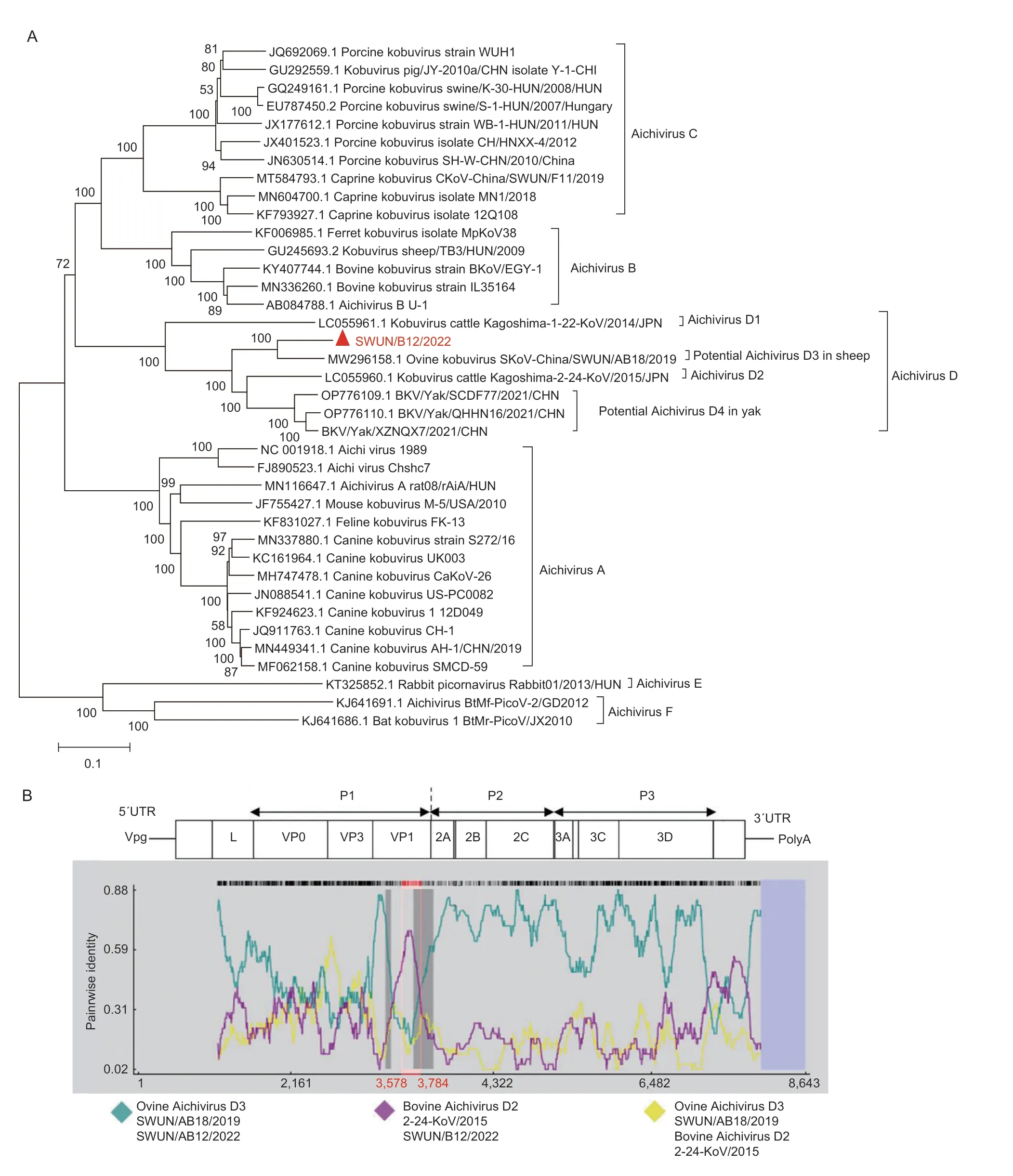
Fig.1 Phylogenetic relationship and recombination analysis of the genome sequences of the novel Aichivirus D strain SWUN/B12/2022.A,phylogenetic relationship of Aichivirus D in this study with representative species from genus Aichivirus based on genome sequences.Sequence alignments and clustering were performed by ClustalW in MEGA 7.0 software.The tree was constructed by the neighbor-joining method with bootstrap values calculated for 1,000 replicates.The red triangle represents the novel Aichivirus D in this study.B,recombination analysis of the genome of the novel Aichivirus D strain SWUN/B12/2022 using RDP 4.0.Plots were constructed using the RDP Method graphical output in RDP4.Red bars schematically indicated the parts of the genome involved in the recombination event.
Furthermore,a recombination event was predicted in the SWUN/B12/2022,based on RDP 4.0 using RDP,GeneConv,Chimaera,SiScan,BootScan and MaxChi methods (recombinant score: 0.549),and the recombination breakpoint was located in the VP1 region at the nt position 3,578-3,784 bp in genome and 533-739 bp in the complete VP1.The putative major parental strain was ovine Aichivirus D,and the possible minor parental strain was bovine Aichivirus D2 (GenBank accession number LC055960) (Fig.1-B),suggesting that the novel Aichivirus D likely originated from ovine Aichivirus D as a result of natural VP1 gene recombination.Notably,ovine Aichivirus D strain may originate from bovine Aichivirus D1 strain (GenBank accession number LC055961) with a recombinant event in the 2C gene (Abiet al.2021).So,recombination events play an important role in the evolution of Aichivirus D (Okitsuet al.2012;Fanet al.2013;Akagamiet al.2017).In fact,recombination events in Kobuvirus,particularly in the VP1,VP3,and 2C genes,have been extensively reported (Okitsuet al.2012;Chen Let al.2013;Fanet al.2013;Akagamiet al.2017;Abiet al.2021).
To further investigate the association between Aichivirus D and goat diarrhea,other 116 goats (aged 2 d to 2 mon) fecal samples (61 diarrheic and 55 non-diarrheic) were collected between June 2021 and January 2022 from 3 farms experiencing diarrhea outbreaks in Sichuan.Using a specific RT-PCR assay developed in this study (Appendix A),the average positive rate of Aichivirus D in diarrheal samples was detected as 55.74% (34/61),significantly higher (P<0.001) than positive rate of 14.55% (8/55) in healthy samples;moreover,the positive rate of Aichivirus D in diarrheal samples from each farm exceeded that of healthy samples (Appendix C).Meanwhile,other common diarrhea-causing pathogens were not detected,except for enterovirus,which had a detection rate of 13.55%.In summary,these results suggest that the novel Aichivrus D is associated with goat diarrhea in the tested farms.Several Kubovirus species have been linked to diarrhea in humans and other animals (Chen Y Set al.2013;Kitajima and Gerba 2015;Yanget al.2015;Abiet al.2022),the results of this study have significant implications for diagnosing and controlling diarrhea in goats.However,given the limited survey scope in this study,further expanded investigations are needed to determine the epidemic characteristics of the virus.
To gain further insights into the molecular characteristics of the P1 region,additional 9 complete P1 (VP0,VP3 and VP1) genes were obtained from Aichivirus D-positive goat fecal samples on 3 different farms using the amplified primers (Appendix A).The VP0 genes were 1,137 bp,encoding 378 amino acid (aa) residues (GenBank accession numbers OP376932-OP376940).The VP3 genes were 669 bp,encoding 223 aa residues (GenBank accession numbers OP376941-OP376949).The VP1 genes were 819 bp,encoding 273 aa residues (GenBank accession numbers OP376950-OP376958).Notably,recombination events identical to that of SWUN/B12/2022 (Fig.1-B) were also observed in the other 9 VP1 genes,which supports the deduction that goat Aichivirus D likely originated from ovine Aichivirus D as a result of natural VP1 gene recombination.
The phylogenetic trees of the VP0,VP3 and VP1 protein sequences indicated that the novel Aichivirus D in this study,along with ovine Aichivirus D,clustered into a large branch (Fig.2-A-C).However,the VP0,VP3 and VP1 protein sequences of the novel goat Aichivirus D strains formed an independent,small branch (Fig.2-AC),indicating that the 10 goat Aichivirus D strains display a unique evolutionary pattern compared to ovine Aichivirus D.

Fig.2 Molecular characteristics of the novel Aichivirus D complete P1 region (VP0,VP3 and VP1 genes).A-C,Aichivirus phylogenetic analysis based on the putative amino acid sequences of the VP0 (A),VP1 (B) and VP3 (C) proteins.Sequence alignments and clustering were performed by ClustalW in MEGA 7.0 software.The tree was constructed by the neighbor-joining method with bootstrap values calculated for 1,000 replicates.The red triangles represent the 10 novel Aichivirus D in this study.D,amino acid variants in the P1 region (VP0,VP3,and VP1 genes) in this study.The figures in the box indicate the amino acid change sites in antigenicity peptide and receptor recognition domain of all the 10 strains in this study compared with the ovine Aichivirus D strain SWUN/AB18/2019 P1 region.
Notable,compared to ovine Aichivirus D,the 10 goat Aichivirus D strains shared 32 unique aa mutations in VP0,14 unique aa mutations in the VP3,and 14 unique aa mutations in VP1 (Fig.2-D),which contribute to the unique evolutionary characteristics of the goat Aichivirus D strains.The VP0 and VP1 protein of kobuvirus are involved in receptor recognition,host range,and antigenic diversity (Chen Y Set al.2013;Zhuet al.2016).Among the 32-aa mutations in VP0,9-aa mutations locate in the potential receptor-binding domain (200-261 aa).In human Aichivirus A,the antigenic epitope (DNSPQPRTTFDYTDNPLPPDTKL) and the receptor motif (PRPPPPLPPLPTP) of VP1 protein have been identified,which locate in 21-43 aa (Chen Y Set al.2013) and in 228-240 aa residues,respectively (Zhuet al.2016).Similar motifs were observed in the goat Aichivirus D in this study,and the antigenic epitope motif and the receptor motif of the VP1 protein are located in 22-44 aa and 227-239 aa residues similar to ovine Aichivirus D (Abiet al.2021).Interestingly,compared to ovine Aichivirus D,the 10 VP1 proteins in this study shared 3 identical aa mutation sites in the conserved antigenic peptide domain of the VP1 protein,which may result in antigenic distinction from sheep Aichivirus D strains;besides,one unique aa mutation was found in the potential receptor-recognition domain of the VP1 protein,this may be a result of Aichivirus D adapting to the host goat,which need to be further confirmed.
In previous studies,we successfully isolated Aichivirus C from goats and Aichivirus D from yaks using Vero cells (Abiet al.2022;Yanet al.2023),so we attempted to isolate Aichivirus D from goats using the same method.A total of 34 positive fecal samples of Aichivirus D from goats were used to viral isolation according to previous reports,however,only 2 samples exhibited noticeable cytopathic effects (CPE) during 3 generations of incubation (Appendix D-1).The 2 samples were verified to be Kobuvirus through RT-PCR,indirect immunofluorescence (IF) assay,and transmission electron microscopy (EM) (Appendix D-2-4).Unfortunately,the viral load of the 2 isolates decreased in the 4th generation of culture and could not be detected in the 5th generation,indicating that virus isolation was unsuccessful.Further efforts are needed to optimize the isolation system for goat Aichivirus D,which is critical for determining the viral pathogenicity.
In summary,this study first identified a novel Aichivirus D from goats,which was most closely related to ovine Aichivirus D.The virus may be originated from ovine Aichivirus D as a result of natural VP1 gene recombination.Moreover,this virus may be associated with diarrhea in goats.These results significantly contribute to our understanding of the host range and genetic evolution of Aichivirus D.
Acknowledgements
This work was sponsored by the Southwest Minzu University Double World-Class Project,China (XM2023014),the Sichuan Veterinary Medicine and Drug Innovation Group of China Agricultural Research System (SCCXTD-2020-18),the Key Laboratory of Veterinary Medicine of Universities of Sichuan Province of China,and the Fundamental Research Funds for the Central Universities,Southwest Minzu University,China (ZYN2023043).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Ethical approval
This study did not involve animal experiments besides the fecal sampling of diarrhea goats that visited farm for clinical treatment;thus no ethics approval was required for this study.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2023.11.041
杂志排行

Journal of Integrative Agriculture的其它文章
- OsNPF3.1,a nitrate,abscisic acid and gibberellin transporter gene,is essential for rice tillering and nitrogen utilization efficiency
- Fine mapping and cloning of the sterility gene Bra2Ms in nonheading Chinese cabbage (Brassica rapa ssp.chinensis)
- Basal defense is enhanced in a wheat cultivar resistant to Fusarium head blight
- Optimized tillage methods increase mechanically transplanted rice yield and reduce the greenhouse gas emissions
- A phenology-based vegetation index for improving ratoon rice mapping using harmonized Landsat and Sentinel-2 data
- Combined application of organic fertilizer and chemical fertilizer alleviates the kernel position effect in summer maize by promoting post-silking nitrogen uptake and dry matter accumulation