ldentification of S-RNase genotype and analysis of its origin and evolutionary patterns in Malus plants
2024-05-13ZhaoLiuYuanGaoKunWangJianrongFengSimiaoSunXiangLuLinWangWenTianGuangyiWangZichenLiQingshanLiLianwenLiDajiangWang
Zhao Liu ,Yuan Gao ,Kun Wang ,Jianrong Feng ,Simiao Sun ,Xiang Lu,Lin WangWen Tian,Guangyi WangZichen LiQingshan Li,Lianwen LiDajiang Wang#
1 Research Institute of Pomology,Chinese Academy of Agricultural Sciences/Key Laboratory of Horticultural Crops Germplasm Resources Utilization,Ministry of Agriculture and Rural Affairs,Xingcheng 125100,China
2 College of Agriculture,Shihezi University/Key Laboratory of Special Fruits and Vegetables Cultivation Physiology and Germplasm Resources Utilization of Xinjiang Production and Construction Corps,Shihezi 832000,China
Abstract Identification of the S genotype of Malus plants will greatly promote the discovery of new genes,the cultivation and production of apple,the breeding of new varieties,and the origin and evolution of self-incompatibility in Malus plants.In this experiment,88 Malus germplasm resources,such as Aihuahong,Xishuhaitang,and Reguanzi,were used as materials.Seven gene-specific primer combinations were used in the genotype identification.PCR amplification using leaf DNA produced a single S-RNase gene fragment in all materials.The results revealed that 70 of the identified materials obtained a complete S-RNase genotype,while only one S-RNase gene was found in 18 of them.Through homology comparison and analysis,13 S-RNase genotypes were obtained: S1S2 (Aihuahong,etc.),S1S28 (Xixian Haitang,etc.),S1S51 (Hebei Pingdinghaitang),S1S3 (Xiangyangcun Daguo,etc.),S2S3 (Zhaiyehaitang,etc.),S3S51 (Xishan 1),S3S28 (Huangselihaerde,etc.),S2S28 (Honghaitang,etc.),S4S28 (Bo 11),S7S28 (Jiuquan Shaguo),S10Se (Dongchengguan 13),S10S21 (Dongxiangjiao) and SeS51 (Xiongyue Haitang).Simultaneously,the frequency of the S gene in the tested materials was analyzed.The findings revealed that different S genes had varying frequencies in Malus resources,as well as varying frequencies between intraspecific and interspecific.S3 had the highest frequency of 68.18%,followed by S1 (42.04%).In addition,the phylogenetic tree and origin evolution analysis revealed that the S gene differentiation was completed prior to the formation of various apple species,that cultivated species also evolved new S genes,and that the S50 gene is the oldest S allele in Malus plants.The S1,S29,and S33 genes in apple-cultivated species,on the other hand,may have originated in M.sieversii,M.hupehensis,and M.kansuensis,respectively.In addition to M.sieversii,M.kansuensis and M.sikkimensis may have also played a role in the origin and evolution of some Chinese apples.
Keywords: Malus,S-RNase genotype,self-incompatibility,origin and evolution
1.lntroduction
Self-incompatibility is a reproductive mechanism that has evolved in plants over time to prevent inbreeding and promote cross-pollination (McClureet al.2006).Plants are classified into two types based on the genetic mechanism of self-incompatibility: sporophytic selfincompatibility (SSI) and gametophytic self-incompatibility (GSI) (Steinet al.1991;de Nettancourt 1997;Takayamaet al.2000;Zhanget al.2006).Apple (Malus domesticaBorkh.) is a typical gametophyte-incompatible plant with self-incompatibility traits regulated by a complex allele at a singleSlocus that belongs to the apple subfamily of the Rosaceae family (Sassaet al.2016).TheSlocus generally contains two types of genes: the styleSgene (S-RNase) and the pollenSgene (SFB/SLF).Only when the pollen carries a differentSgene from its own style does the pollen tube germinate normally into the ovary,completing fertilization (Kao and Tsukamoto 2004;Xuet al.2008;de Franceschiet al.2012;Heet al.2021).Since theS-RNasegene was first identified in tobacco plants of the Solanaceae family (East and Mangelsdorf 1925),a number of Rosaceae styleSgenotypes have been identified (Chenet al.2013;Liang 2019;Huet al.2021;Heet al.2022;Weiet al.2022).In practice,self-incompatibility limits apple yield improvement and quality improvement,and suitable pollinator trees must be reasonably deployed.Different pollinator varieties have been shown to have a significant impact on apple fruit quality,primarily improving fruit set rate,fruit shape index,anthocyanin and soluble sugar content,and aroma substances (Zhanget al.2018;Zhao and Zhang 2019).Therefore,identifyingSgenotypes of differentMalusresources can help guide the appropriate allocation of pollinating trees and provide a solid foundation for improving yield and quality.
Kobelet al.(1939) first identified theSgenotype of varieties such as ‘Ralls’ by cross-pollination and pollen tube observation methods,and identifying 11 differentS-RNasealleles.Following that,many researchers have used isoelectric focusing electrophoresis (IEF),two-dimensional polyacrylamide gel electrophoresis (2D-PAGE),and non-equilibrium pH gradient electrophoresis (NEPHGE) to differentiate nucleotide sequence differences in the appleS-RNasegene to identify theS-RNasegene (Sassaet al.1994;Verdoodtet al.1998;Boskovic and Tobuut 1999;Matsumotoet al.2003;Kimet al.2008).With the development of molecular biology techniques in recent years,Sgenotype identification techniques based on PCR or PCR digestion techniques have successfully identified 1,024Malusresources and established a database of appleSgenotypes (Longet al.2010a,b;Liet al.2011,2017;Zhanget al.2013;Dinget al.2018).
China is the world’s largest genetic center for apple plants,with a wealth of original apple species and types (Gaoet al.2020a,2021),as well as some foreign varieties and preserved germplasm resources.However,theSgenotypes of some apple varieties and germplasm resources preserved in China have not yet been fully identified.In addition,the identification of appleS-RNasegenes has long been mostly focused on cultivars,and further identification and mining ofSgenotypes from various wild resources and ancient cultivars is still required (Liet al.2017).
Therefore,88Malusplants (including cultivars,ancient cultivated varieties and wild resources) were used as test material materials in this experiment to identifyS-RNasealleles in different apple resources using seven pairs of apple-specific primers,providing reference and theoretical basis for enriching the appleSgenotype database,rational selection of pollinating varieties,efficient genetic breeding,and the origin and evolution ofSgenes.
2.Materials and methods
2.1.Plant materials
Young apple leaves ofMalus-tested resources were collected in May 2022 at the National Repository of Apple and Pear Germplasm Resources (Xingcheng),Research Institute of Pomology,Chinese Academy of Agricultural Sciences.Samples were immediately frozen in liquid nitrogen and individually labeled for cryopreservation at -80°C (Table 1).
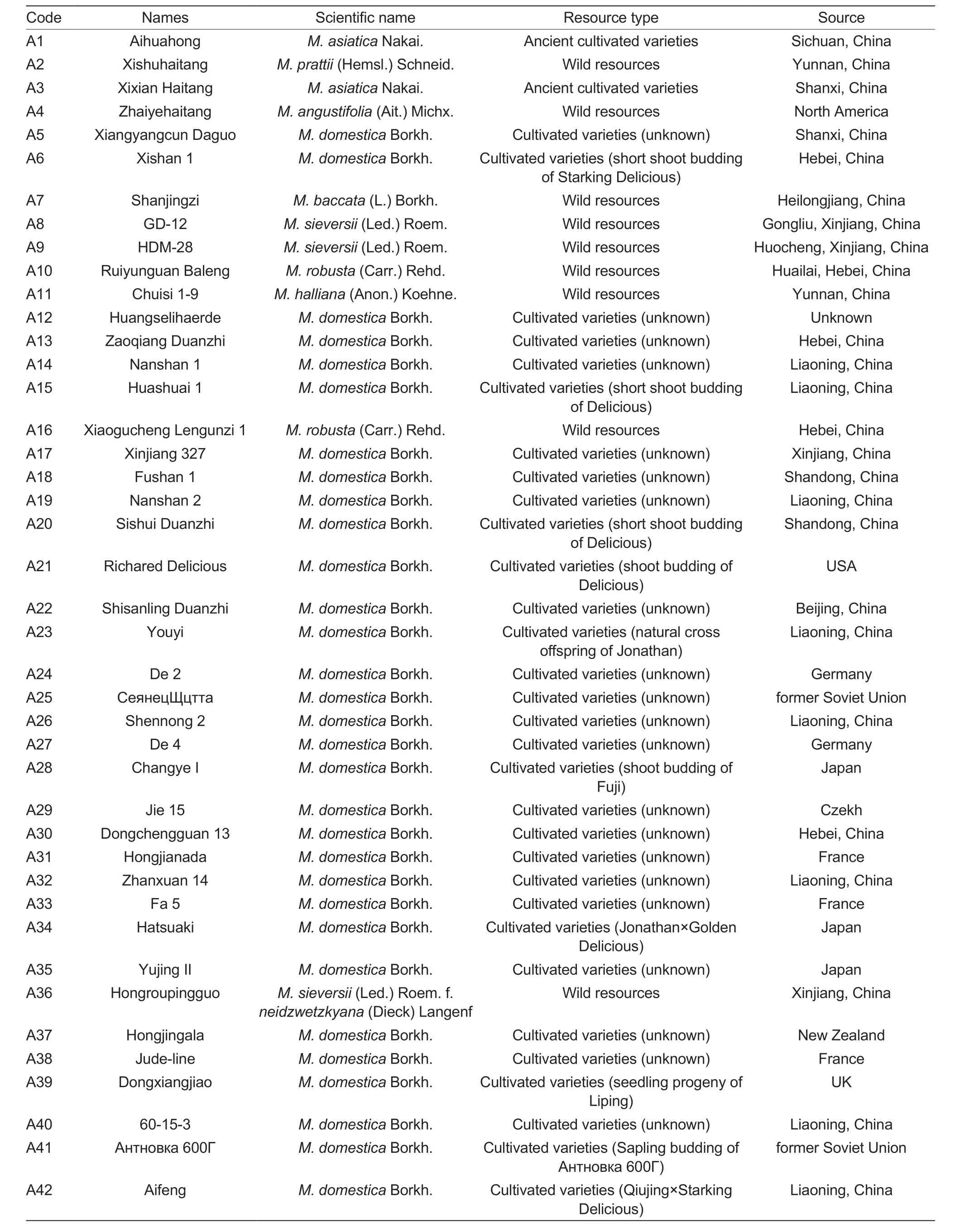
Table 1 Test materials of 88 Malus plants
2.2.DNA extraction
The 88Malusplants in Table 1 were used to extract leaf DNA using the novel plant genomic DNA extraction kit DP320 (Tiangen,Beijing,China).DNA integrity and concentration were determined using 1% agarose gel electrophoresis and the Thermo NANODrop 2000 nucleic acid protein meter.DNA samples were stored at -20°C for backup.
2.3.PCR amplification of the S-RNase gene
Seven pairs of primers (Beijing LiuheHuada Gene Technology Company,Beijing,China) -S-1F+S-1R,S-2F+S-2R,S-3F+S-3R,S-4F+S-4R,S-7F+S-7R,S-19F+S-19R and S-24F+S-24R -were chosen for this experiment based on the known literature on the reportedS-RNasegenes in apple.
PCR amplification was performed using leaf genomic DNA as a template,and 25 μL of PCR reaction system was used: 12.5 μL of 2×TaqPCR MasterMix (TaKaRa,Dalian,China),0.5 μL of each upstream and downstream primers at 0.5 μmol L-1,1 μL of diluted DNA (50 ngμL-1),and ddH2O water comprised 25 μL.The reaction procedure involved pre-denaturation at 94°C for 3 min,denaturation at 94°C for 30 s,extension at 72°C for 1 min and at 72°C for 5 min after 35 cycles,with the specific primer annealing temperatures shown in Table 2.After the PCR reaction was completed,5 μL of the amplification product was taken and separated by electrophoresis on a 1% agarose gel.Then,5 μL of DL2000 DNA marker was used as a reference for the size of the target fragment to detect the presence or absence of the PCR amplification product and the size of the target fragment,and a gel imaging system was used to take photographs after the agarose gel electrophoresis.
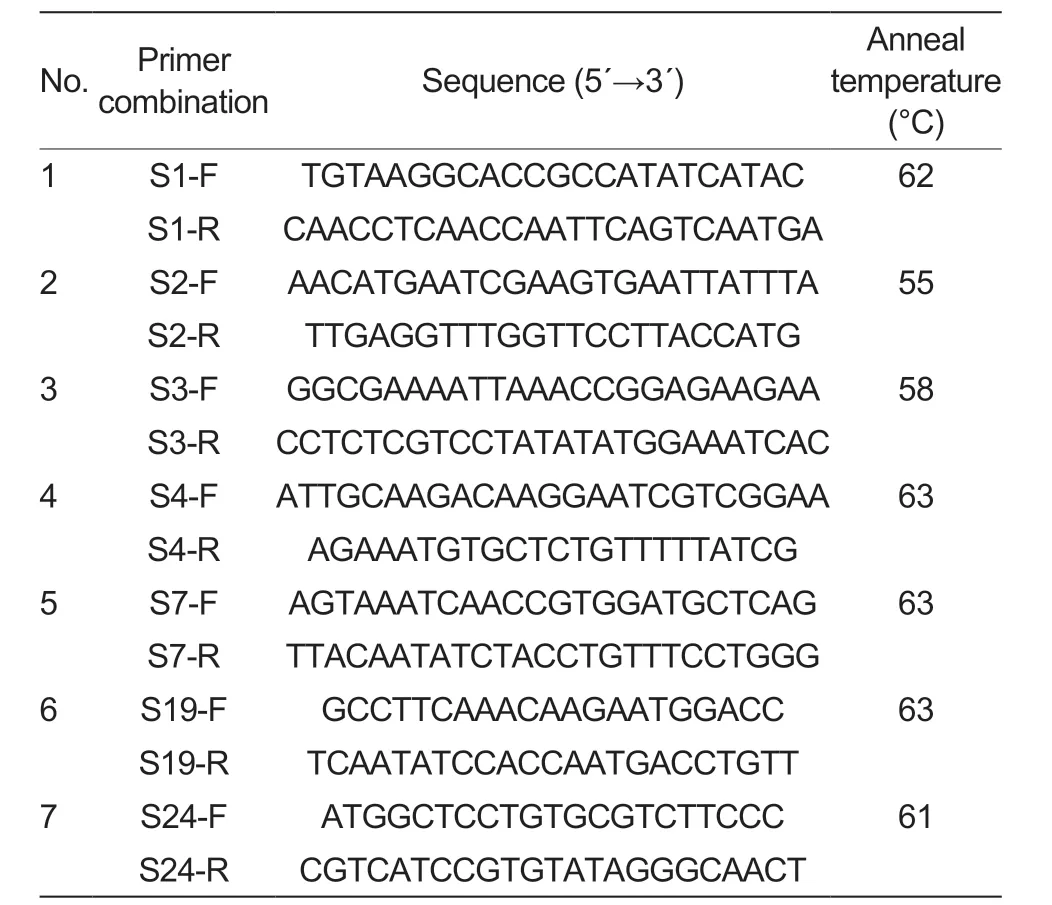
Table 2 Sequences of consensus primer combinations1)
2.4.Cloning and sequencing of the S-RNase gene
The target bands of the PCR products were recovered and purified using the DNA Recovery and Purification Kit (Tiangen,Beijing,China),and the purified target fragments were ligated into the pEASY-Blunt-Simple cloning vector and introduced into Trans5α receptor cells at 42°C for single-colony positive clone selection.At least six single colonies were selected for detection of the target bands,and at least three single colonies containing the target bands were selected for sequencing;all sequencing results were performed by Beijing Liuhe Huada Gene Technology Company (Beijing,China).
2.5.Data analysis
The nucleotide sequences obtained in this experiment were subjected to BLAST homology comparison analysiswith known sequences in the NCBI database to determine that the sequencedS-RNasegenes (https://www.ncbi.nlm.nih.gov/genbank/).A nucleotide sequence homology greater than 98% could be considered as the sameSgenotype (Zenget al.2014).The full-length sequences of the registeredS-RNasegenes ofMalusof Rosaceae were also selected from the database and compared separately using the neighbor-joining method to plot and construct a phylogenetic tree with MEGA11.0 software.
3.Results
3.1.Specific amplification of the S-RNase gene
The results of seven pairs ofS-RNasespecific primers amplification revealed that 70 of the 88 materials all amplified two distinct target bands,while the remaining 18 materials all amplified only one target band (Fig.1).The fragment lengths of all the target bands were concentrated between 317 and 755 bp (Table 3).When all the nucleotide sequences from the test were compared for homology using GenBank,the BLAST findings revealed that the genes with the highest nucleotide sequence homology to the test’sS-RNasegenes were all members of the Rosaceae family’s genus apple,and the amplified fragments corresponded to 11S-RNasegenes.S1gene fragment sizes were 755,741,731,726,and 720 bp (e.g.,White Pippin,Aihuahong,Xishuhaitang,etc.);S2gene fragment sizes were 542,534,523,517,and 508 bp (e.g.,Ruiyunguan Baleng,Zhaiyehaitang,Chuisi 1-9,etc.);S3gene fragment sizes were 353,340,330,327,and 317 bp (e.g.,Xiangyangcun Daguo,Aifeng,De 4,etc.);theS4gene fragment size was 560 bp (e.g.,Bo 11);theS7gene fragment size was 442 bp (e.g.,Jiuquan Shaguo);theS10gene fragment size was 538 bp (e.g.,Dongchengguan 13,Dongxiangjiao);S21gene fragment size was 374 bp (e.g.,Dongxiangjiao);theS24gene fragment size was 726 bp (e.g.,Yichun Shaguo,Wumahezhenhuawei Shaguo);theS28gene fragment sizes were 523,515,502,and 492 bp (e.g.,Nanshan 1,Xixian Haitang,Zaoqiang Duanzhi,etc.);theS51gene fragment sizes were 511 and 506 bp (e.g.,Xishan 1,Xiongyue Haitang,etc.);and theSegene fragment size was 369 bp (e.g.,Dongchengguan 13,etc.).

Fig.1 The specific amplified fragments of the S gene from 88 Malus plants.
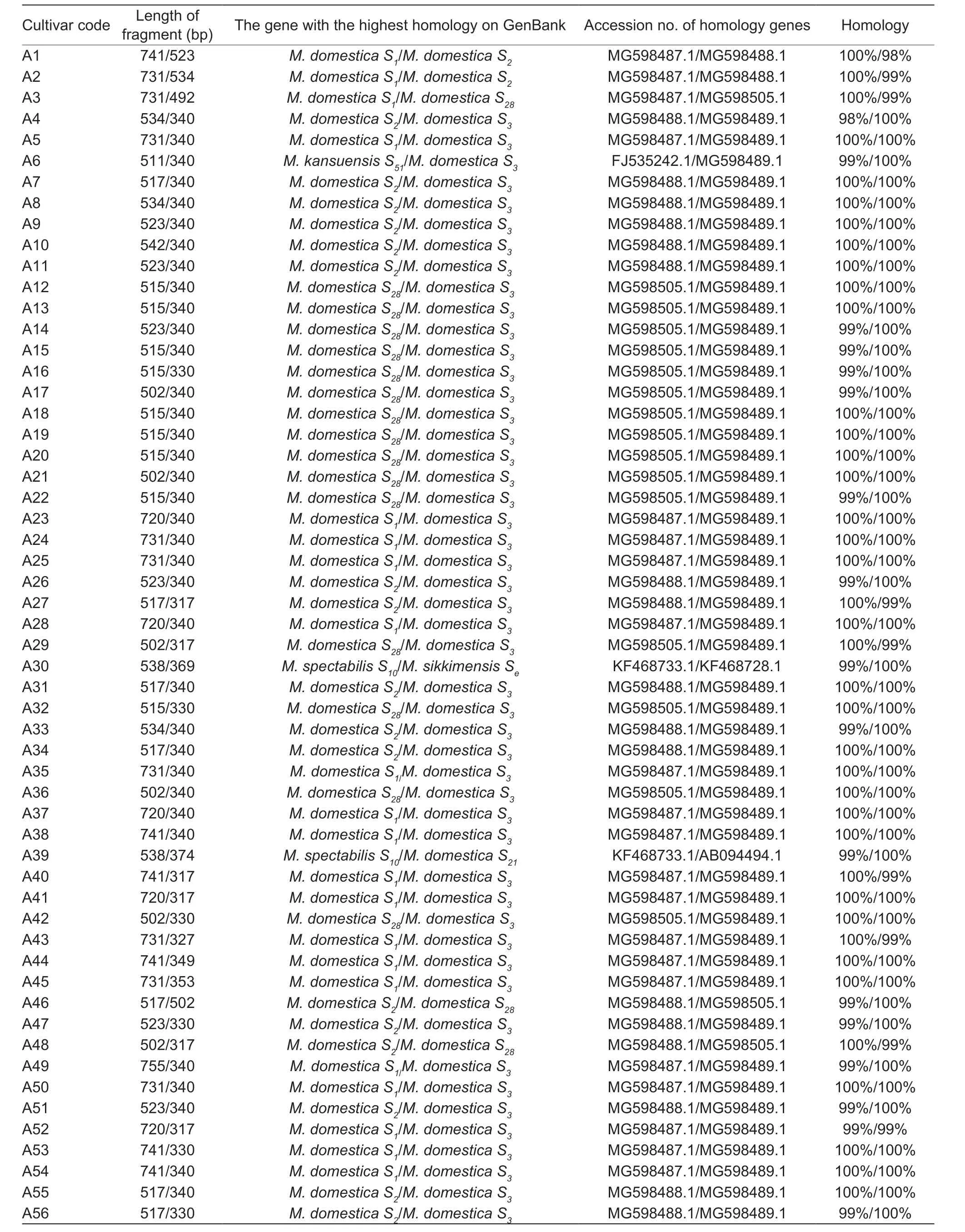
Table 3 Comparison results of the gene fragments from 88 Malus plants
3.2.ldentification of the S-RNase genes
TheS-RNasegenotypes of the 88 participatingMalusplants were identified using the results of a homology comparison of the bands in GenBank.Several of the test materials had the sameSgenotype.For instance,theSgenotypes of Aihuahong,Xishuhaitang,Regunzi,and Luanzhuang Shaguo were allS1S2;theSgenotypes of Xixian Haitang,Jie 9,and Sankuaishi Haitang were allS1S28;theSgenotypes of 21 materials,including Xiangyangcun Daguo,Youyi,and De 2,were allS1S3;theSgenotypes of 16 materials,including Zhaiyehaitang and Shanjingzi,were allS2S3;and theSgenotypes of 17 materials,including Huangselihaerde,Zaoqiang Duanzhi,and Nanshan 1,were allS3S28.However,Hebei Pingdinghaitang,Xishan 1,and Xiongyue Haitang all had one identicalS51gene;Honghaitang,Huamei,Bo 11,and Jiuquan Shaguo all had one identicalS28gene;and Dongchengguan 13 and Dongxiangjiao had one identicalS10gene.In addition,18 materials were identified with only oneS-RNase,with three specimens,including Youfangcun Shanjingzi,having only theS2gene identified;five specimens,including Rixin Shaguo 1,having only theS3gene identified;eight specimens,including Guangmiangunzi,having only theS1gene identified;and two specimens,including Wumahezhenhuawei Shaguo,having only theS24gene identified (Table 4).

Table 4 S-genotype of 88 Malus plants
3.3.Statistical analysis of the frequency of S-RNase genes
The change in gene frequency is the foundation of evolution.The frequency of genes is influenced by various factors,including gene mutations,gene recombination,natural selection,migration,and genetic drift.The frequency of genes is constantly changing,leading to the continuous development and evolution of organisms (Lv 2015).Therefore,analyzing the frequency changes of theSgene is beneficial for understanding the genetic differences and evolution among differentMalusplants.The results of the 88 test materials revealed the involvement of 11 differentS-RNasegenes,with the frequency ofSgenes varying significantly (Fig.2-A).TheS3gene was amplified in 60 materials with the highest frequency of 68.18%,followed by theS1gene amplified in 37 materials with a frequency of 42.04%,and theS2andS28genes appeared with nearly the same frequency of 28.40 and 27.27%,respectively.TheS4,S7,S10,S21,S24,S51,andSegenes each appeared between one and three times,with frequencies ranging from 1.13 to 3.40%.
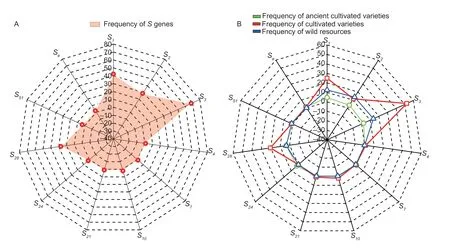
Fig.2 Analysis of the frequency of S genes.A,S gene frequency of 88 Malus plants.B,S gene frequency of different Malus plants.
In the total test materials,10 of the ancient cultivated varieties wereMalus asiatica,51 were cultivated varieties,and 27 were wild resources.The analysis revealed that the frequency ofSgenes varied depending on the resource type.S1,S2,S3,S28,andS51were theSgenes shared by the ancient cultivated varieties,cultivated varieties,and wild resources.The frequency of distribution of these five genes in cultivated varieties was higher than in old cultivars and wild resources.The ancient cultivar Xiongyue Haitang and the cultivar Dongchengguan 13 shared theSegene,whereas theS4,S10,andS21genes were only found in cultivated varieties (Bo 11,Dongchengguan 13,and Dongxiangjiang).TheS7andS24genes were only found in ancient cultivated varieties (Jiuquan Shaguo,Wumahezhenhuawei Shaguo,and Yichun Shaguo),and there was no statistically significant difference in their frequency of occurrence (Fig.2-B).
The 11 apple species involved in this experiment includedM.domesticaBorkh.,M.asiaticaNakai.,M.prattii(Hemsl.) Schneid.,M.robusta(Carr.) Rehd.,M.angustifolia(Ait.) Michx.,M.baccata(L.) Borkh.,M.sieversii(Led.) Roem.,M.halliana(Anon.) Koehne.,M.sieversii(Led.) Roem.f.neidzwetzkyana(Dieck) Langenf,M.sylvestrisMill.,andM.orientalisUglitz.The analysis revealed that the distribution frequency of theS-RNasegene was also different between species and within species.The analysis ofS-RNasegene frequencies in different apple species revealed that theS2andS3genes were distributed in nineMalusspecies,and bothS2andS3had the highest distribution frequencies in cultivars (11.36 and 52.27%,respectively),followed by theS1andS28genes in four species.The frequency ofthe S1gene was higher in cultivars (25.00%) andM.robusta(11.36%),while the frequency of theS28gene was highest in cultivars (20.45%),with little difference in the frequency distribution in the remaining three species.The analysis ofS-RNasegene frequencies within the same species ofMalusresources revealed that there were nine differentSgenes in the cultivars,and the frequency distribution ofSgenes varied greatly,with theS3gene having the highest frequency distribution of 52.27%,followed by theS1gene (25.00%),and theS4,S21,S51,andSegenes having the lowest frequency distribution of 1.13%.There were eight differentSgenes in theM.asiatica;theS1gene had the highest frequency of 4.54%,followed by theS2(3.40%) andS28(3.40%) genes.Among the five differentSgenes inM.robusta,theS1gene had the highest frequency of 11.36%.There were no significant differences in the frequency ofSgenes among and within species in these eight species due to the relatively small number ofMalusresources in the eight species selected for this study,such asM.prattii,M.angustifolia,andM.baccata(Fig.3).
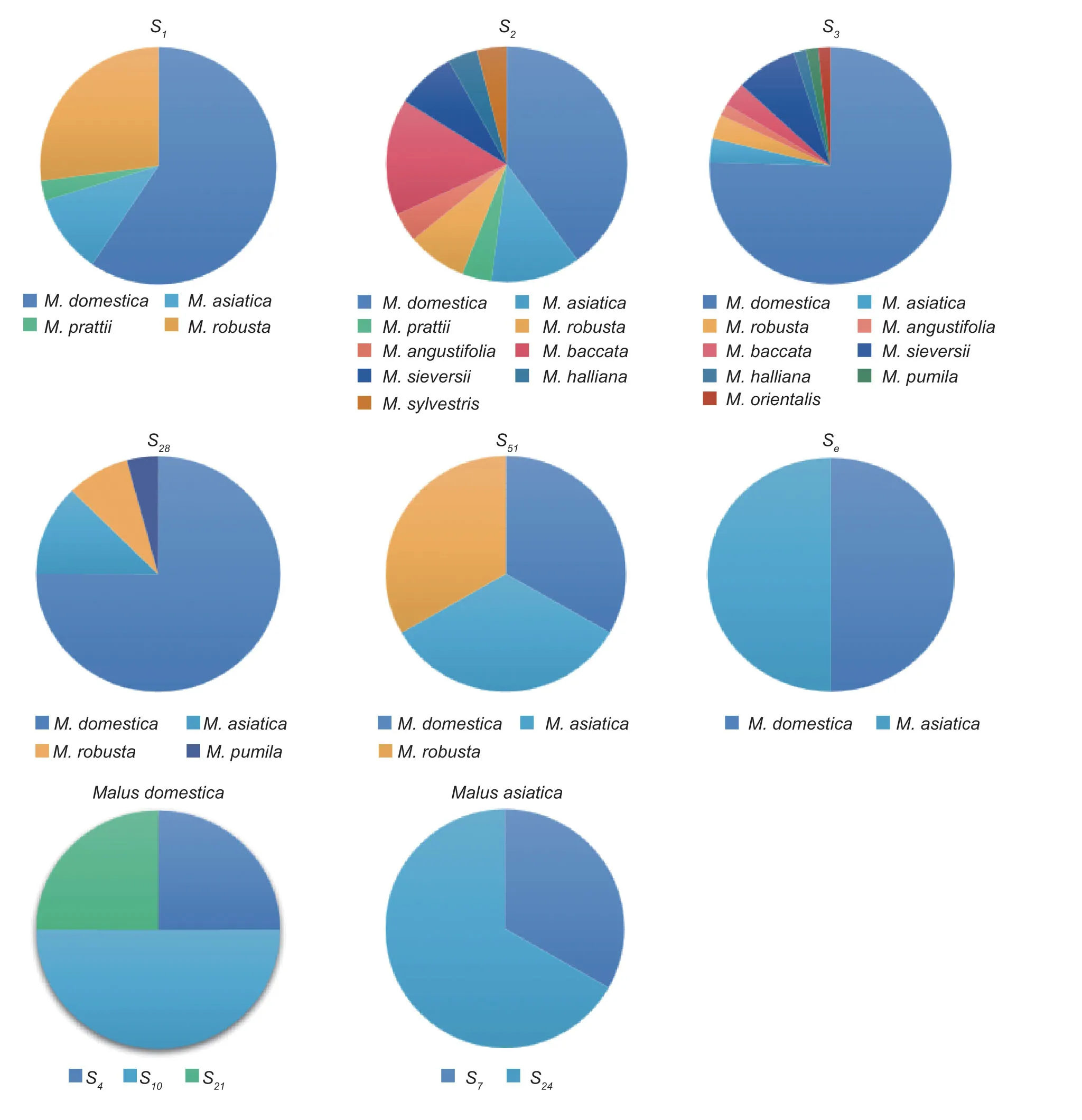
Fig.3 S-RNase gene frequency among and within species of different Malus plants.
3.4.Phylogenetic analysis of S-RNase genes
To investigate the phylogenetic relationships ofS-RNasegenes inMalusplants,the NCBI database was searched for registered appleS-RNasegenes,and the 11S-RNasegene sequences obtained from this experiment were used to construct a phylogenetic tree using MEGA11.0 software.The genusMaluswas chosen to includeM.sieversii,M.domestica,M.micromalus,M.sylvestris,M.orientalis,M.spectabilis,M.rockii,M.fusca,M.hupehensis,M.mandshurica,M.baccata,M.prunifolia,M.sikkimensis,M.angustifolia,M.toringoides,andM.kansuensis,comprising 94S-RNasegenes from 16 species (Fig.4).
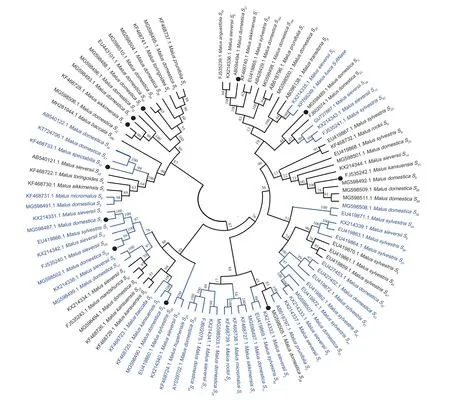
Fig.4 Phylogenetic analysis of the S-RNase gene in Malus plants.● represents the genes obtained in this study;the blue area represents a very close genetic relationship.The data in the figure,from inside to outside,are S-RNase gene registration numbers,Latin names of species,and S-RNase genes.
TheS-RNasegenes obtained in this experiment were on different branches,withS1being related toM.sieversii S1andM.sylvestris S1;S4being related toM.kansuensis S5andM.baccata S5;S24being related toM.sieversii S49;andS10being most closely related toM.domestica S41,S34,andS39b.For the analysis of theS-RNasegene sequences ofMalusplants obtained from NCBI,it could be seen thatM.fusca S-RNasewas related toM.sieversii S5.M.domestica S39andM.sylvestris S39;M.sylvestris S50andM.sieversii S13;M.sieversii S9andM.sylvestris S38andS37;M.domestica S33,andM.sieversii S3,andM.sylvestris S41;andM.prunifolia S2andM.sylvestris S7;M.domestica S11andM.sikkimensis S1;M.micromalus SfandM.rockii Sg;M.domestica S29andM.hupehensis S29;M.sylvestris S46andM.sieversii S10;andM.domestica S17andM.sieversii S8were found to be the closest relatives to each other.In addition,the study found that the uniform distribution ofS-RNasegenes from differentMalusspecies in the evolutionary tree did not show a species classification,nor did it show obvious intraspecific homology higher than interspecific homology across the whole phylogenetic tree.Therefore,the analysis suggests thatS-RNasegenes differentiation should have been completed prior to the formation of various species ofMalus.
3.5.Analysis of the origin and evolution of S-RNase genes
This experiment used the NCBI database to retrieve theSallele ofMalusthat had been registered and had the exact origin in order to further analyze the relationship between the origin and evolution of theS-RNasegene inMalus.According to the study,40Salleles were identified inM.domesticaand 56Salleles were identified in 16 species of wild resource types.TheSgenes identified in wild species were mostly concentrated inM.sylvestrisandM.sieversii.The numbers ofSgenes identified in 14 wild species,includingM.sikkimensisandM.baccata,were small,ranging between one and four.However,the correspondingSalleles had not been identified in the other 15 wild species,includingM.trilobata,M.doumen,andM.xiaojinensis(Fig.5).

Fig.5 Origin and evolution of the S-RNase genes in Malus plants.
Malusplants were classified into six groups based on origin and evolution: the Duoshenghaitang group,Sanlieyehaitang group,Huaqiupingguo group,Lvpingguo group,Shanjingzi group,and Apple group.OnlyM.tschonoskiiin the Duoshenghaitang group had an identifiedS50gene,whileM.doumenandM.mellianahad no identified correspondingSallele.Malus trilobatahad only one species of the Sanlieyehaitang group,and itsSgene had not been identified.There were 13 species in the Huaqiupingguo group.Malus kansuensis(S5,S6,S33,S51),M.transitoria(Sg´),M.toringoides(Sa,Se),M.fusca(S),andM.micromalus(Sf,S8) had identifiedSalleles,respectively,and the other eight species,includingM.florentina,M.prattii,M.yunnanensis,andM.honanensis,hadn’t identified their correspondingSalleles.OnlyM.angustifoliain the Lvpingguo group had an identifiedS48gene,while theSgene had not been identified in the four species ofM.ioensis,M.coronaria,M.glancescens,andM.glabrafa.There were six species in the group of Shanjingzi,and theSgene ofM.sikkimensis(S1,S7,St,Se),M.baccata(S5,S39b),M.hupehensis(S29),M.halliana(S2,S3),M.rockii(Sg,S9),andM.mandshurica(S52) had been identified.The Apple group was mainly composed ofM.sylvestris,M.orientalis,andM.sieversii.Only theS49gene was identified inM.orientalis.Moreover 18Sgenes,such asS36-S41andS44-S47,were identified inM.sylvestris,and theS1-S14,S33,S48,andS49genes were identified inM.sieversii.
4.Discussion
Although the appleSgene database (including 1,024Malusplants) has been applied in practice (Liet al.2017),it primarily contains theSgenotype of a large number of apple cultivars,and theSgenotype of ancient cultivars,varieties,and wild resources has not been widely identified.China’s ancient cultivars and wild resources are not only rich in resources,but also have strong resistance and adaptability to local tastes,and they have played an important role in fruit diet history.These resources are important stress-resistance and special flavor-breeding parents.On the premise that fruit trees are located up and down the mountains and use the four wastelands to develop new fruit orchards,identifying theirSgenotype is critical.In the gametophytic selfincompatibility type,they will still show self-incompatibility,even if different varieties pollinate each other with the sameSgenotype (Takayama and Isogai 2005).The study in this experiment found that Luanzhuang Shaguo (S1S2),Xixian Haitang (S1S28),Honghaitang (S2S28),Jiuquan Shaguo (S7S2),and Xiongyue Haitang (SeS51) had differentSgenotypes despite being ancient cultivar varieties,indicating that these materials had very different genetic backgrounds and were relatively rich in diversity;they therefore had great potential for exploitation.In addition,many of the 88 test materials identified had the sameSgenotype.For example,theSgenotypes of Xiangyangcun Daguo,Youyi,and De 2 wereS1S3;theSgenotypes for Huangselihaerde,Zaoqiang Duanzhi,and Nanshan 1 wereS3S28;and theSgenotypes of Shennong 2,De 4,and Hongjianada wereS2S3.If crossbreeding is required in the future,such varieties with the sameSgenotype should not be used as parents of each other and should avoid being pollinators of each other in production.Only oneS-RNasefragment was amplified from the 18Malusresources in this study.This phenomenon was suggested to be caused by the fact that someS-RNasegenes inMalusplants contain long introns,which reduce the efficiency of PCR amplification (Broothaertset al.1995).Alternatively,these materials may be pureS-RNasegenotypes,resulting in only oneS-RNasegene.
Although several specificSalleles have been cloned inMalusplants,there are 15 commonSgenes,includingS1-S7,S9,S10,S16,S19,S20,S22,S23,S24,andS26.The remainingSgenes only appear in specific resource types (Longet al.2010b).In this study,theSgenes identified by the test materials involved 11Sgenes,and the frequency of their occurrence in differentMalusresources was analyzed.The identification results ofSgenes revealed thatS1,S2,S3,andS28genes were distributed in apple cultivars,ancient cultivated varieties,and wild resources,and these genes appeared more frequently in cultivars than other types.The reason for this phenomenon could be that in the past 30 years of apple cross-breeding,some varieties with good economic characteristics,such as Golden Delicious,Fuji,Ralls,Delicious,Starking Delicious,and Hanfu,have frequently been used as hybrid parents (Liet al.2020).TheSgenotype of Ralls (S1S2),Delicious (S9S28),Golden Delicious (S2S3),Fuji (S1S9),Gala (S2S5),Hanfu (S1S9),and Starking Delicious (S9S19) have been clearly identified by the predecessors (Hoyet al.2006;Longet al.2010a,b;Liet al.2011).The analysis found that theS1,S2,S3,andS28genes contained in these commonly used parents were consistent with the high-frequencySgenes identified in the experiment,whereas theS5,S9,andS19gene were not identified in the cultivated varieties in this experiment.Liet al.(2011) summarized the results of various research units at home and abroad on appleSalleles andSgenotype identification,discovering that among theSgenotypes of more than 500 apple-cultivated varieties that were analyzed accurately,the frequencies ofS5,S9,andS19genes were lower than those ofS1,S2,andS3genes.Therefore,it was speculated that theseSalleles may not be contained in the offspring of Fuji,Gala,and Starking Delicious as the female or male parents in the subsequent cross-breeding,resulting in the high frequency ofS1,S2,S3,andS28genes in apple cultivars varieties.TheS4,S7,S10,S21,S24,S51,andSegenes identified in this experiment only appeared 1-2 times in different wild resource types with a low frequency.S51andSegenes,for example,were found in Xiongyue Haitang,andS24genes were found in Yichun Shaguo.The analysis suggests that the low frequency ofSalleles may be due to the fact that some resources were only preserved as germplasm resources,and these ancient cultivated varieties and wild resources had not been used for breeding purposes.It was also possible that the number of apple-cultivated varieties chosen in this experiment was large,while the number of resources chosen from nine species such asM.prattii,M.angustifolia,andM.baccatawas relatively small,resulting in an incomplete identification of theSgene species and thus the low frequency of these genes.
With regard to the order of the origin and evolution ofMalusplants,some scholars have considered that the Duoshenghaitang group and the Sanlieyehaitang group were the most primitive species based on the detection of phenolic substances in the leaves and cortex and the classification of plant morphology (Williamet al.1982).Scholars have also considered that theM.yunnanensissystem andM.kansuensissystem,both of which originated in China,were all members of the Huaqiupingguo group,and then Lvpingguo group,Shanjingzi group,and Apple group (Williamet al.1982;Yu 1984;Li 1999).The clustering and origin evolution ofSgenes were investigated in this study based on the order of the origin and evolution ofMalusresources.This study showed thatM.domestica S1andM.sieversii S1were clustered together,as wereM.domestica S29andM.hupehensis S29,M.domestica S26andM.fusca S26,M.domestica S33andM.kansuensis S33,andM.domestica S39andM.sylvestris S39.This shows that genetic relationship was very close.Malus hupehensisbelonged to the Shanjingzi group andM.kansuensisbelonged to the Huaqiupingguo group,both of which were older than the Apple group.Therefore,it is believed that theS29andS33genes originated from the Shanjingzi group and Huaqiupingguo group and then gradually evolved into the Apple group.A recent study revealed that a newS-RNasegene (S67-RNase) was identified from four native Chinese pear,exhibiting high similarity withM.domestica S32(Heet al.2022).In addition,studies have revealed thatM.domestica S4andM.domestica S9gene is on the same evolutionary branch asPyrus pyrifolia S49andP.pyrifolia S53(Jianget al.2017).These results further indicate a close genetic relationship betweenMalusandPyrus,and supporting the hypothesis that the existence ofS-RNasepredates the species formation betweenMalusandPyrus.Malus sieversiiandM.sylvestrishave been proved to participate in the evolution process ofM.domestica(Velascoet al.2010;Duan 2017).This study’s clustering results also demonstrated a close genetic relationship between theM.sieversii,M.sylvestris,and cultivated apple.Moreover,theS1gene ofM.domesticawas highly homologous toM.sieversii S1.It is believed that theS1gene inM.domesticamay have originated fromM.sieversii.Furthermore,Sunet al.(2020) concluded thatM.sylvestrisacted as the maternal lineage in the evolution ofM.domesticathrough genome assembly and pangenome sequencing of Gala,M.sieversii,andM.sylvestris.The analysis ofSgenes fromM.domesticaandM.sylvestrisin this experiment revealed that cultivarS41had high homology withM.sylvestris S41,implying that theS41gene may have originated fromM.sylvestrisand then gradually participated in the evolution ofM.domestica.According to the conventional wisdom,Chinese apple evolved fromM.sieversii,which is primarily composed of Mianpingguo and its related species (Li 2001),includingM.asiatica,M.robusta,M.micromalus,M.prunifolia,andM.doumen.In theory,Chinese apple should have a high homology with theSgenes ofM.sieversii.In this study,theSgene analysis ofM.asiatica(S1,S2,S3,S7,S24,S28,S51,Se),M.robusta(S1,S2,S3,S28,S51),andM.prunifolia(S1,S2,Se),which were closely related to Chinese apple,showed that theS1,S51,andSegenes inM.asiatica,M.robusta,andM.prunifoliahad high homology withM.sieversii(S1),M.kansuensis(S51),andM.sikkimensis(Se).Gaoet al.(2020b) suggested that in the origin evolution,part of the germplasm of Chinese apples had the genetic background ofM.sieversii,M.baccata,andM.robusta,and their formation was more complicated than that of cultivated apples.TheSgene analysis suggests that,in addition toM.sieversii,M.kansuensisandM.sikkimensismay have also been involved in the evolution of the origin of some Chinese apples.However,the evolutionary relationship between Chinese apples,M.sieversiiand otherM.species needs to be further investigated.
With the origin and evolution order,theSgene ofMaluswas either lost or newly evolved.TheS50gene was found to exist simultaneously inM.tschonoskii,M.sylvestris,andM.domesticain this study.M.tschonoskiiwas an ancient primitive species that belonged to the Duoshenghaitang group.It can be considered thatS50gene was the oldestSallele ofMalus.According to the evolutionary order,theSgenes identified in the Huaqiupingguo group were mainly followed,such as theSaandSegenes ofM.toringoides,theSg´ genes ofM.transitoria,and then the genes identified in the Lvpingguo group (S48),the Shanjingzi group (S39b,Sg,etc.),and the Apple group (S46,S58,etc.) (Fig.6).The analysis suggests that theS49gene,which existed inM.sylvestris,M.orientalis,andM.sieversiibut had not been identified inM.domestica,may be the preference or gene mutation ofSgene in the genetic process,resulting in gene deletion or a newSgene phenomenon.In addition,the analysis ofSgene in wild resources found that theSgene only existed in their own species,with little gene exchange,and that theSgene in most wild resources did not inherit into cultivated apple with the origin and evolution.However,someSgenes,such asSkbandS58,had recently evolved in cultivated apples,indicating that gene mutations or genetic hitchhiking effects occurred during theSgene’s origin and evolution.
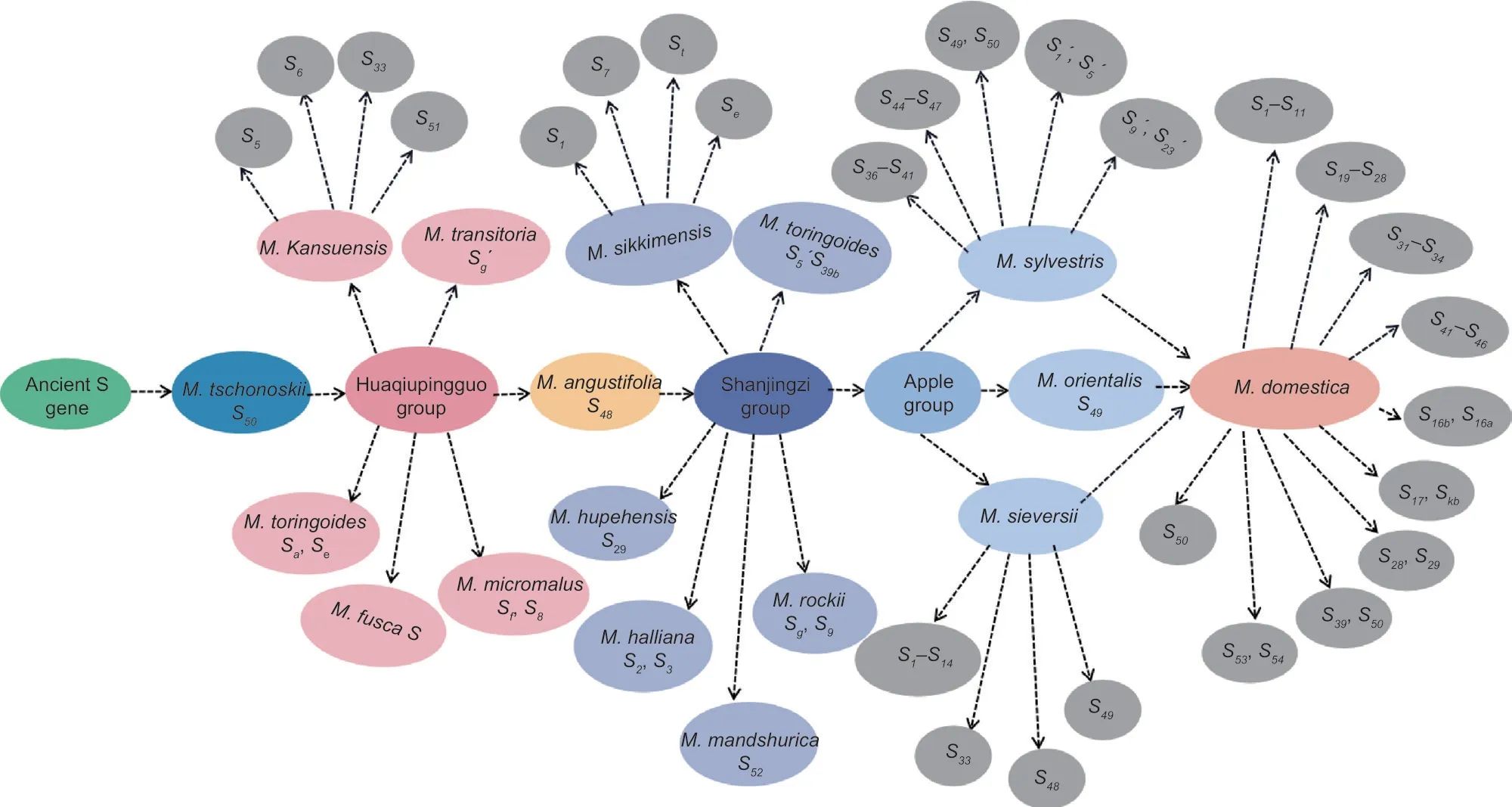
Fig.6 Speculation on the possible evolutionary route of the S-RNase gene in Malus plants.
In addition,this study found that someSgenes inMalusplants have homonym phenomenon according to the NCBI database retrieval statistics (Table 5).For example,theM.domestica S1andM.sikkimensis S1,M.sieversii S1andM.sikkimensis S1,M.domestica S2andM.sieversii S2,M.domestica S3andM.sieversii S3,andM.domestica S5andM.baccata S5gene have the sameSgene name,but the homology difference is large,which is considered to be the phenomenon of homonymous foreign matter.Liet al.(2011) sorted and renamed theSgene of the known apple cultivar at that time,so it was suggested to rearrange theSgene naming of wild resources after systematically sorting and summarizing theSgene that appeared in the wild resource types and cultivated apples.

Table 5 Homonymous and suggestions of the S gene in Malus plants
5.Conclusion
In this study,we identified theSgenotype of 88 test materials using seven pairs ofS-RNasegene specific primers inMalusgermplasm resources.The results revealed that 70 of the identified materials obtained a completeS-RNasegenotype,while only oneS-RNasegene was found in 18 of them.Meanwhile,the frequency of theSgene revealed that differentSgenes had varying frequencies inMalusresources,as well as varying frequencies between intraspecific and interspecific.Further,the phylogenetic tree and origin evolution analysis revealed that theS50gene is the oldestSallele inMalusplants,while theS1,S29,andS33genes in applecultivated species may have originated fromM.sieversii,M.hupehensis,andM.kansuensis,respectively.In addition,M.kansuensisandM.sikkimensismay have also played a role in the origin and evolution of some Chinese apples.
Acknowledgements
This research was financially supported by the Agricultural Science and Technology Innovation Program (CAASASTIP-2021-RIP-02).
Declaration of competing interest
The authors declare that they have no conflict of interest.
杂志排行

Journal of Integrative Agriculture的其它文章
- OsNPF3.1,a nitrate,abscisic acid and gibberellin transporter gene,is essential for rice tillering and nitrogen utilization efficiency
- Fine mapping and cloning of the sterility gene Bra2Ms in nonheading Chinese cabbage (Brassica rapa ssp.chinensis)
- Basal defense is enhanced in a wheat cultivar resistant to Fusarium head blight
- Optimized tillage methods increase mechanically transplanted rice yield and reduce the greenhouse gas emissions
- A phenology-based vegetation index for improving ratoon rice mapping using harmonized Landsat and Sentinel-2 data
- Combined application of organic fertilizer and chemical fertilizer alleviates the kernel position effect in summer maize by promoting post-silking nitrogen uptake and dry matter accumulation