伴单克隆B淋巴细胞和单克隆浆细胞增殖的AITL 1例
2024-04-30徐腾飞刘金立朱永村
徐腾飞 刘金立 朱永村
1威海市立医院检验科,威海 264200;2威海市立医院病理科,威海 264200
病例资料
患者,女,73岁,2015年1月27日因“查体发现淋巴结肿大10个月余,加重半年”于威海市立医院入院治疗。既往有“甲状腺功能减退症、左眼孔源性视网膜脱离手术、阑尾炎手术、肝胆管结石”病史。体检示:全身皮肤广泛大小不等斑丘疹;双侧颈部、腋窝、腹股沟可触及肿大的淋巴结,质韧,无触痛,活动可;颜面部、眼睑、双下肢轻度水肿;双肺呼吸音清,心律齐,腹部无压痛及反跳痛,肝脾肋下未触及。血常规结果:白细胞计数8.56×109/L,淋巴细胞百分数6.42%,中性粒细胞百分数76.41%,单核细胞百分数13.3%,血红蛋白106 g/L,血小板计数43×109/L。C反应蛋白59 mg/L,血沉16 mm/h,降钙素原0.61 µg/L。体液免疫结果:血清Kappa轻链1.65 g/L、Lambda轻链10.75 g/L,β2-微球蛋白4.16 mg/L。血清电泳分析:白蛋白61.4 g/L,M蛋白7.2%。免疫固定电泳免疫球蛋白(immunoglobulin,Ig)G Lambda型见图1A。甲状腺功能结果:游离三碘甲状腺原氨酸1.94 pmol/L,甲状腺球蛋白<0.2 µg/L,抗甲状腺过氧化物酶抗体37.1 U/ml。凝血功能、肿瘤标志物基本正常,抗核抗体谱正常。EB病毒检测阳性。MYD88-L265P基因突变阴性。外周血T淋巴细胞亚群占比:CD3+53.2%,CD3+CD4+35.2%,CD3+CD8+17.2%,CD3-CD19+1.8%,CD3-CD(16/56)+23.2%;另可见CD3-CD4+CD8-异常T细胞占淋巴细胞17.3%。骨髓形态学结果:增生明显活跃,大部分淋巴细胞可见颗粒,浆细胞比例增高,余未见异常细胞(图1B)。骨髓免疫分型结果:骨髓中可见一群CD3-CD4+CD8-异常T淋巴细胞,占有核细胞的1.42%,表达CD10、程序性死亡受体1(PD-1),不除外血管免疫母细胞性T细胞淋巴瘤(angioimmunoblastic T-cell lymphoma,AITL);另可见异常B淋巴细胞及浆细胞,占有核细胞的比例分别为3.73%、11.7%(图2)。淋巴结病理结果:(左腋窝)内梭形细胞增生,淋巴结结构破坏,散在不规则滤泡,滤泡间细胞成分较杂,伴淋巴细胞、浆细胞、嗜酸性粒细胞浸润,可见异型淋巴细胞增生,胞体小至中等大,胞核轻度不规则;免疫组化提示增生梭形细胞为肌源性和滤泡树突细胞源性。免疫组化结果:CD3+、CD2+、CD5+、CD7-、CD4+、CD8-、CD10+、PD1+、BCL6+、CXCL13(少量+)、CD21(FDC+)、Ki-67(30%~40%)、CD20-、CD79a-(见图3)。结合患者临床表现、骨髓穿刺、淋巴结病理组织化学结果,患者诊断为AITL伴B淋巴细胞及浆细胞异常增殖。告知患者家属病情后建议化疗,家属同意使用EPOCH联合CD20及CD38化疗方案。2015年7月15日至2016年12月16日,共进行8个疗程后得到完全缓解,2020年1月22日,因复发严重入院,家属最终放弃治疗。

图1 1例伴单克隆B细胞和单克隆浆细胞增殖的血管免疫母细胞性T细胞淋巴瘤患者免疫电泳及骨髓形态学检查结果。A:免疫电泳可见明显免疫球蛋白G和λ区带;B:骨髓中可见异常淋巴细胞和浆细胞(瑞氏-吉姆萨染色 ×1 000)

图2 1例伴单克隆B淋巴细胞和单克隆浆细胞增殖的血管免疫母细胞性T细胞淋巴瘤患者骨髓多参数流式免疫分型结果。A、B、C、D:异常T淋巴细胞(黑色群)免疫表型CD3-CD4+CD8-CD5+CD10+PD-1+;E、F:单克隆B淋巴细胞(紫色群)轻链cLambda+;G、H:单克隆浆细胞(蓝色群)轻链cLambda+
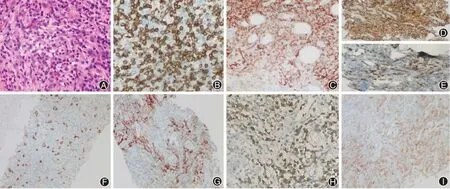
图3 1例伴单克隆B淋巴细胞和单克隆浆细胞增殖的血管免疫母细胞性T细胞淋巴瘤患者的淋巴结活组织病理检查和免疫组织化学检查结果。A:淋巴结活检显示正常结构被小到中型淋巴样细胞破坏,细胞质清晰,细胞核轻度不规则,高内皮小静脉明显增生;可见类似Reed-Stemberg(RS)细胞的嗜酸性核仁多核细胞(HE ×400);B:CD3阳性(EliVision ×400);C:CD10阳性(EliVision ×100);D:CD4阳性(EliVision ×400);E:CD8阴性(EliVision ×400);F:CD20阳性(EliVision ×100);G:CD21阳性(EliVision ×100);H:CXCL13阳性(EliVision ×400);I:PD-1阳性(EliVision ×100)
讨 论
AITL是一种来源于滤泡辅助T细胞(follicular helper T cell,TFH)的肿瘤,发病中位年龄50~60岁,男性略有优势[1-2]。大多数患者临床表现为晚期疾病,主要症状为全身性淋巴结肿大、皮疹、肝脾肿大,约70%患者可累及骨髓,淋巴结外侵袭部位以肺、胃肠道多见[3]。此外,某些患者还存在由免疫功能失调导致的自身免疫性疾病,可表现为高γ球蛋白血症及自身免疫性溶血性贫血[4]。本病例为73岁女性患者,初诊时发热、盗汗,体表淋巴结多处肿大,全身皮肤广泛大小不等的斑丘疹。实验室检查贫血、淋巴细胞减低、乳酸脱氢酶升高,但自身免疫性抗体谱正常,疾病后期出现肝脾肿大,并累及骨髓及胸腔。
AITL病理学可见恶性细胞呈多态炎性细胞背景,由反应性B淋巴细胞、免疫B淋巴母细胞、组织细胞、嗜酸性粒细胞和浆细胞组成。高内皮小静脉和滤泡树突状细胞增殖是AITL特异性结构[5]。免疫组织化学上,肿瘤细胞除表达泛T细胞抗原CD3、CD2、CD5外,还表达多种TFH相关抗原,如PD-1、CD10、BCL6、CXCL13、ICOS、SAP和CXCR5[6]。近年来,流式细胞术在AITL初诊及其多种继发改变的监测诊断中起重要作用。AITL流式免疫表型分析:常见异常T细胞,比例不高;mCD3减少甚至缺失,表达CD4而不表达CD8;CD10的异常表达是AITL特异性表型[7]。Leval[8]指出,AITL肿瘤细胞可过表达CXCL13,对PD-1和可诱导共刺激分子(ICOS)较敏感,对CXCL13和CD10较特异。Yabe等[9]研究表明,sCD3-/CD4+异常细胞群是晚期AITL患者特征性免疫表型。本例患者外周血及骨髓中均检测到异常T细胞,流式免疫表型为mCD3-CD4+CD8-CD10+,占有核细胞的比例不高,易被忽略。
目前,AITL发病机制的探究已取得重大进展。全外显子组和靶向测序研究已确定了一系列基因突变,主要包括编码表观遗传调控因子的甲基胞嘧啶双加氧酶2(methylcytosine dioxygenase 2,TET2)、异柠檬酸脱氢酶2(isocitrate dehydrogenase 2,IDH2)、DNA甲基化转移酶3A(DNA methyltransferase 3A,DNMT3A)和Ras同源基因家族成员A(Ras homolog gene family,member A,RHOA)[10]。TET2突变见于约80%的AITL,多数病例表现出2个或多个TET2突变,TET2酶功能丧失可影响细胞发育,一个关键过程可能是幼稚CD4+细胞阶段,TET2下调将增强BCL6基因表达并启动向TFH倾斜分化[11]。RHOA突变仅限于PD-1+富集具有TFH特征的细胞,可能通过独立机制增强信号传导,指定TFH谱系,维持TFH表型,并引发自身免疫反应[12]。TET2和RHOA突变都可以增强TFH与幼稚CD4+细胞的分化。IDH2突变是一种独特的AITL亚型,发生率为30%~40%。IDH2突变可影响酶活性,使a-酮戊二酸还原为2-羟基戊二酸的量增加,羟基戊二酸可抑制组蛋白去甲基化酶,从而异常调节基因转录过程[13]。IDH2突变还可降低T细胞增殖和TCR信号传导。此外,IDH2突变的AITL可表现出骨髓受累,细胞学特征为中大型透明肿瘤细胞,TFH细胞标志物升高,滤泡辅助树突状细胞网络扩大;这些结果表明,IDH2可能会影响AITL的肿瘤微环境。为了证实此观点,Leca等[14]对AITL队列进行了基因富集比对分析,发现IDH2突变的样本显示与TFH细胞、血管生成、代谢、缺氧相关的基因富集;与IDH2野生型相比,IDH2突变型AITL肿瘤显示干扰素调节因子4和缺氧诱导因子染色增强,血管数量增加,证实了上述TFH和血管生成的富集基因特征。与急性髓系白血病中的互斥存在方式不同,IDH2突变的AITL几乎总伴随TET2突变。小鼠模型验证中,TET2和IDH2突变必须在TFH细胞中结合,这与人类AITL中TET2和IDH2突变的共存相一致。与IDH2突变小鼠相比,TET2突变小鼠胸腺亚群或CD3+T细胞总数差异均无统计学意义(均P>0.05),但双重突变导致脾脏中CD4+和CD8+初始T细胞减少,CD4+和CD8+记忆细胞增加。此外,TET2和IDH2联合突变对TFH细胞增殖没有影响,但会改变其表型和功能;TET2和IDH2联合突变通常显示组蛋白甲基化水平显著增加及全基因组启动子区域高甲基化;因此,组蛋白去甲基化可能是AITL的重要靶点。DNMT3A在AITL突变发生频率为20%~38%,TFH淋巴瘤中的大多数DNMT3A突变与TET2突变同时发生;DNMT3A突变参与克隆造血,但它们是否直接参与TFH淋巴瘤的分化仍不清楚[15]。基因突变与病理形态学具有一定的相关性。IDH2突变可出现反应性、裸露的生发中心和滤泡间扩张;RHOA可出现多个退行性“燃尽”的生发中心;无IDH2和RHOA突变的淋巴结结构完全消失,没有残留的生发中心[16]。
AITL伴随的免疫母细胞化B细胞增殖和EB病毒感染的永生化B细胞克隆性扩增可能是由于AITL出现的免疫功能失调导致EB病毒感染或再激活刺激所致。Leca等[14]研究表明,遗传因素在驱动B淋巴细胞增殖中也发挥重要作用。在T细胞依赖性免疫应答过程中,正常TFH和B细胞之间可相互作用产生多种共刺激信号,TFH细胞中的TET2突变可导致周围正常B细胞改变对共刺激信号受体的表达,从而驱动克隆B细胞增殖,导致侵袭性B细胞淋巴瘤的发生。当TFH细胞携带TET2和IDH2联合突变时,生发中心B细胞和浆细胞增加,AITL样疾病出现。IDH2抑制剂可纠正肿瘤性TFH细胞因子的分泌,减少B细胞增殖[17]。以上结果表明,遗传突变对B细胞克隆性增殖具有重要影响。AITL中存在的浆细胞通常是多克隆的,单克隆浆细胞增殖较罕见。约80%病例中单克隆浆细胞EB病毒阴性,这表明EB病毒再激活不是主要的病理来源。本病例中,免疫组织化学显示骨髓浆细胞EB病毒阴性,与先前报道结果一致。事实上,EB病毒感染的记忆B细胞在分化为产生抗体的浆细胞后从潜伏状态转变为裂解状态[18]。因此,AITL中克隆性浆细胞扩增的病理学机制与B细胞淋巴增殖的机制可能不同,有必要进一步了解AITL中克隆性浆细胞增殖的潜在机制。本例患者检测到异常T细胞,比例不高,反而异常B淋巴细胞和浆细胞比例较高,易被误诊为成熟B细胞淋巴瘤和异常浆细胞疾病,单克隆浆细胞增殖是本病例的重要特点,较罕见。单克隆B淋巴细胞及浆细胞形态、免疫表型与正常细胞无异,只是单纯轻链克隆性产生变化,与淋巴浆细胞性淋巴瘤表型相似,但MYD88-L265P基因突变及IgM蛋白的检测结果可除外淋巴浆细胞性淋巴瘤。
研究表明,患者年龄、美国东部肿瘤协作组体能状态评分、β2-微球蛋白等可有效预测AITL患者预后[19]。AITL一线化疗方案CHOP疗效欠佳、复发率高,建议年轻、复发的AITL患者进行大剂量化疗,随后进行自体干细胞移植。临床药物开发在免疫治疗方向探索较多,但仍不成熟。近年来,几种分子靶向药物已在日本获批用于治疗复发或难治性外周T细胞淋巴瘤,但尚未发布关于AITL药物的具体指导意见;研究显示,每种药物对AITL的总有效率为8%~30%,与其他外周T细胞淋巴瘤相似[20]。本例患者年龄较大,不易移植,故采用EPOCH联合CD20及CD38化疗方案;8个疗程结束后达到完全缓解,效果较好,但维持3年后,患者因复发较严重入院,家属最终放弃治疗。
利益冲突所有作者均声明不存在利益冲突
作者贡献声明徐腾飞:论文撰写,数据采集;刘金立:数据采集,论文修改;朱永村:研究指导
