中药白矾与伪品铵明矾的中红外光谱快速鉴别△
2024-04-29朱雨欣董玲宋平顺倪琳陈建波
朱雨欣,董玲,宋平顺,倪琳*,陈建波*
1.北京中医药大学 生命科学学院,北京 102488;2.甘肃省药品检验研究院,甘肃 兰州 730070
中药白矾是由硫酸盐类矿物明矾石提炼制成,其中十二水合硫酸铝钾 [KAl(SO4)2·12H2O] 质量分数不应低于99.0%[1]111-112。2016 年,中国食品药品检定研究院收集来自全国范围内饮片生产企业与市场的159 批白矾样品,发现其中125 批样品的含氮量偏高、铵盐检查项不符合规定[2]。此后的全国药品抽检中白矾(包括其煅制品枯矾)的不合格率也超过30%[3-4]。白矾不仅自身是一味常用中药,也是半夏、天南星、白附子等的炮制辅料。因此,白矾掺伪会导致较多品种中药饮片与中成药的质量隐患,必须建立准确有效的白矾真伪鉴别方法。
当前市售白矾伪品主要是铵明矾[2-3],其成分为十二水合硫酸铝铵 [NH4Al(SO4)2·12H2O],因外观性状与白矾正品基本一致而难以分辨。《中华人民共和国药典》(以下简称《中国药典》)2020 年版将白矾质量标准中的铵盐检查法修订为氮测定法(通则0704 第二法或第三法,无需消解),规定白矾药材和饮片中含铵盐以总氮(N)计不超过0.3%[1]111-112,从而可以准确识别铵明矾掺伪的白矾样品。但是,《中国药典》2020 年版规定的半微量氮测定法(通则0704第二法)和定氮仪法(通则0704第三法)的操作繁琐,需要使用硼酸溶液、氢氧化钠溶液、硫酸滴定液、滴定指示剂等化学试剂,检测过程耗时长、成本高、环境污染大。在国家全力实现“双碳”目标的战略背景下,中药鉴别方法也应朝向更加简便、快速、费用低、绿色的方向发展,提高检测效率,降低检测成本,减少检测过程的碳排放。
已有研究探索了使用扫描电镜、X 射线衍射、近红外(NIR)光谱、拉曼(Raman)光谱等分析技术对白矾、枯矾、铵明矾及其煅制品进行检测的可行性[5-8]。扫描电镜与X 射线衍射设备要求较高,不适用于日常检验。NIR 光谱与Raman光谱操作简单,但是邻近谱峰重叠导致白矾与铵明矾的差异特征峰不够直观[8]。另外,NIR光谱与Raman光谱并非《中国药典》2020 年版使用的化学药或中药鉴别标准方法,采用这2 种技术新建白矾鉴别标准可能增加检验机构和企业的设备成本。相比之下,《中国药典》2020 年版中称为“红外分光光度法”[9]的中红外(MIR)光谱具有与NIR 光谱、Raman 光谱相同的简便、快速、费用低、绿色等优势。此外,MIR 光谱可以同时表征中药样品有机成分和无机成分的整体组成[10],既能靶向检测已知掺假物质,又能非靶向筛查未知掺假物质。此外,MIR 光谱作为国内外药典普遍使用的化学药鉴别标准方法,也是《中国药典》2020 年版规定的中药石膏鉴别标准方法[1]98,所需设备在相关机构与企业的普及率很高,可以显著降低新检测方法的开发与应用成本。
1 材料
1.1 仪器与试药
BT25S 型电子分析天平(德国Sartorius 公司);配有Platinum ATR 附件的Vertex 70 型傅立叶变换红外光谱仪(德国Bruker公司);配有UATR Two 附件的Spectrum 2 型傅立叶变换红外光谱仪(美国PerkinElmer 公司);内置衰减ATR 附件的IT2000 型傅立叶变换红外光谱仪(北京鉴知技术有限公司);Spectrum 2 与IT2000 型光谱仪主要用于样品MIR 测试结果重现性考察,其余测试工作均使用Vertex 70型光谱仪完成,3 台光谱仪放置于不同机构的实验室。
十二水合硫酸铝钾(批号:Z16A10Y95447,纯度:99.8%)、十二水合硫酸铝铵(批号:Z26J10Y9 1549,纯度:99.5%)均购自上海阿拉丁生化科技股份有限公司。
1.2 样品
23 批白矾样品来源见表1,经北京中医药大学中药学院王晶娟教授鉴定,其中11 批(A01~A11)为正品白矾[KAl(SO4)2·12H2O]、12 批(B01~B12)为伪品铵明矾 [NH4Al(SO4)2·12H2O]。

表1 白矾样品来源信息
2 方法
2.1 样品处理
每批样品取50 g 左右粉碎,取粉末直接进行MIR光谱测试。
不同粒度白矾和铵明矾样品制备:将白矾(A01)与铵明矾(B01)粉碎后依次通过二号筛、四号筛、六号筛、九号筛,得到不同粒度的样品粉末。
白矾和铵明矾模拟混合物制备:6 个白矾样品、6 个铵矾样品分别配对(A01+B01、A02+B07、A03+B02、A04+B09、A05+B10、A06+B11);每组配对的白矾和铵明矾样品各自粉碎,过四号筛后按比例混匀,获得铵明矾质量分数分别为5%、10%、20%的混合物(A01+B01 组合中增加铵明矾质量分数为1%的混合物)。
2.2 MIR光谱测试与数据处理
每个样品测试前先后用纯水、无水乙醇清洁ATR 附件的内反射晶体表面,溶剂挥干后测量背景光谱;然后取样品粉末适量置于内反射晶体表面,用ATR 附件自带压杆使样品与晶体表面紧密接触,测量样品光谱。MIR 光谱测量范围4000~400 cm-1(IT2000型光谱仪为4000~500 cm-1),光谱分辨率为4 cm-1,单张光谱累计扫描32 次,以透过率为纵坐标记录样品原始光谱。
使用Spectrum v10.4.3 软件对样品原始光谱依次进行如下处理:1)通过插值功能将所有光谱横坐标间隔调整为1 cm-1;2)将光谱纵坐标由透过率转为吸光度(A);3)对光谱进行ATR 校正,接触因子设为0;4)对光谱进行自动基线校正;5)对光谱纵坐标进行归一化,使2000~800 cm-1区域内最高A为1、最低A为0;6)以1500、1300 cm-1为基线两端参考点,计算1500~1300 cm-1区域内整体峰面积、1450~1430 cm-1区域内最强峰高度,然后用Microsoft Excel 2016 软件进行峰面积与峰高度统计分析。
3 结果与讨论
3.1 白矾与铵明矾的MIR光谱测试方法考察
为了提高样品均匀度与代表性,固体样品通常粉碎后进行MIR 光谱测试,而粉末粒度是影响ATR法测量结果的潜在因素。如图1 所示,粒度越小的白矾与铵明矾MIR 光谱中水分子O-H 伸缩振动峰(3000 cm-1附近宽强峰)、弯曲振动峰(1600 cm-1附近中等强峰)的强度越低,可能是因为样品粒度越小时粉碎过程中水分损失越多。但是,粉末粒度没有显著改变各吸收峰位置,几乎不影响1436 cm-1附近吸收峰是否存在的判断结果。因此,为了简化前处理过程,白矾或铵明矾样品粉碎后无需过筛,直接使用ATR法测试MIR光谱即可。
1.3 统计分析 使用 SPSS 16.0和 Microsoft Office Excel 2007进行统计学分析,主要采用均数、标准差、全距、总数、百分比进行统计描述,采用t检验。

图1 不同粒度的白矾与铵明矾的MIR光谱
白矾(A01)和铵明矾(B01)在同一台MIR光谱仪(Vertex 70)上连续6 次测量结果见图2。水分子O-H 伸缩振动峰(3000 cm-1附近宽强峰)强度有一定变化,可能是因为测量过程的结晶水挥发散失。如表2、表3 所示,白矾(A01)和铵明矾(B01)重复测试的峰位置RSD≤0.3%,说明本研究采用的MIR光谱测试方法重复性良好。
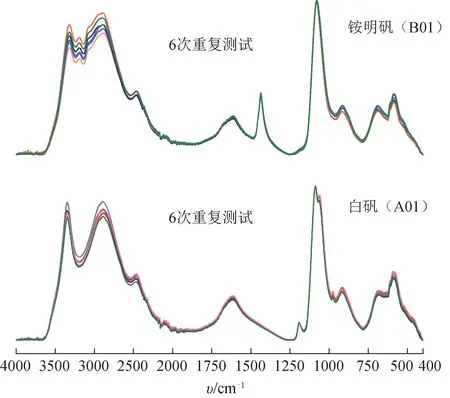
图2 同一仪器重复测试的白矾与铵明矾的MIR光谱
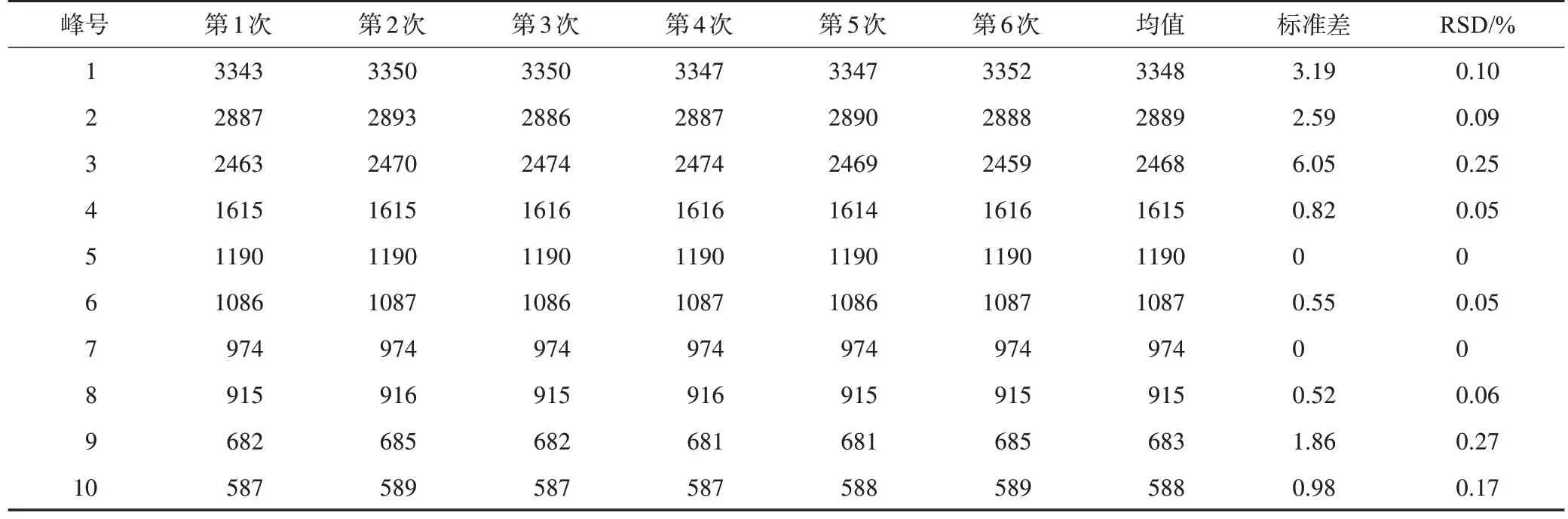
表2 同一仪器重复测试的白矾(A01)MIR光谱吸收峰波数 cm-1
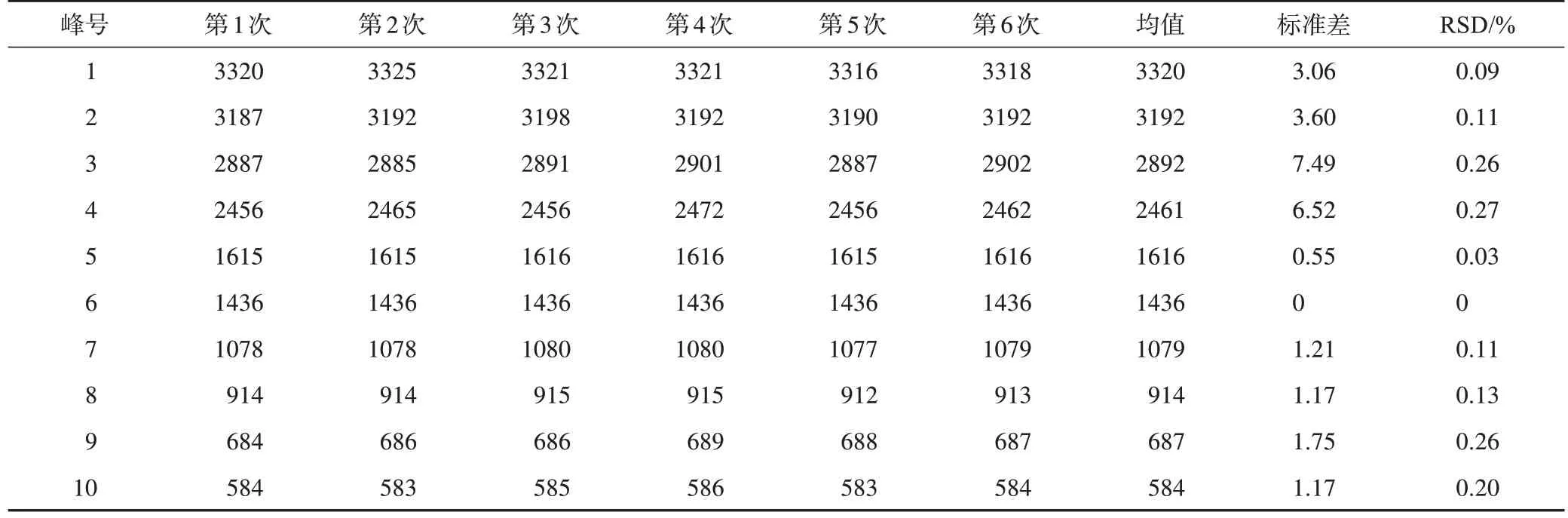
表3 同一仪器重复测试的铵明矾(B01)MIR光谱吸收峰波数 cm-1
白矾(A01)和铵明矾(B01)在不同实验室不同MIR 光谱仪上的测量结果见图3。水分子O-H 伸缩振动峰(3000 cm-1附近宽强峰)、弯曲振动峰(1600 cm-1附近中等强峰)强度有较为明显的差异,可能是因为不同测试环境中测量过程的结晶水挥发散失程度不同。如表4 所示,白矾(A01)和铵明矾(B01)在不同实验室不同MIR 光谱仪上测试的峰位置RSD≤1.0%,说明本研究采用的MIR光谱测试方法重现性良好。
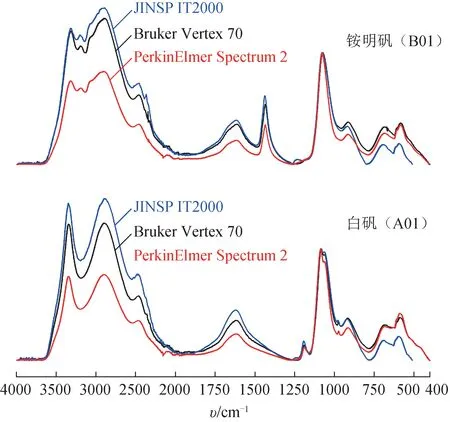
图3 不同实验室不同仪器测试的白矾与铵明矾的MIR光谱

表4 不同实验室不同仪器测试的白矾(A01)与铵明矾(B01)MIR光谱吸收峰波数 cm-1
3.2 白矾与铵明矾的MIR光谱解析与差异吸收峰
如图4 所示,十二水合硫酸铝钾MIR 光谱中3346、2895、2469 cm-1为水分子O-H 伸缩振动峰,1615 cm-1为水分子O-H 弯曲振动峰,1190、1083、1061、974、916 cm-1为硫酸根S-O 伸缩振动峰,684、589 cm-1为硫酸根S-O 弯曲振动峰;十二水合硫酸铝铵MIR光谱中3318、2887、2460 cm-1为水分子O-H 伸缩振动峰,1622 cm-1为水分子O-H 弯曲振动峰,1187、1067、909 cm-1为硫酸根S-O 伸缩振动峰,679、577 cm-1为硫酸根S-O 弯曲振动峰,3204、3075 cm-1为铵离子N-H 伸缩振动峰,1435 cm-1为铵离子N-H弯曲振动峰。
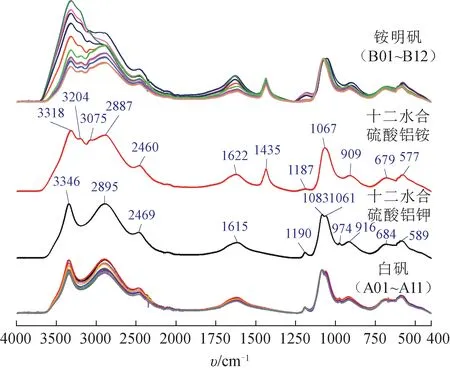
图4 市售真、伪白矾样品与对应化合物的MIR光谱
比较十二水合硫酸铝钾与十二水合硫酸铝铵的MIR 光谱可知,两者明显的差异是铵离子相关吸收峰。但是,3204、3075 cm-1附近的铵离子N-H 伸缩振动峰与水分子O-H 伸缩振动峰重叠严重而不易识别,所以1435 cm-1处的铵离子N-H 弯曲振动峰是辨别十二水合硫酸铝钾与十二水合硫酸铝铵的关键特征峰。白矾与十二水合硫酸铝钾的MIR 光谱基本一致,而铵明矾与十二水合硫酸铝铵的MIR 光谱基本一致,所以铵离子N-H 弯曲振动峰也是区分白矾与铵明矾的关键特征峰。可能因为含有不同比例的水分和杂质,本研究所收集的铵明矾样品的N-H 弯曲振动峰在(1436±1)cm-1区域有微小变动。
为了尽可能减少1600 cm-1附近O-H 弯曲振动峰对1436 cm-1附近N-H 弯曲振动峰强度计算的影响,本研究选择1500、1300 cm-12 点建立局部基线。如图5 所示,分别以峰面积(1500~1300 cm-1区域光谱在局部基线以上的积分面积)、峰高度(1450~1430 cm-1区域光谱最高点到局部基线的距离)作为N-H 弯曲振动峰强度的量化参数。白矾没有N-H 弯曲振动峰,而O-H 弯曲振动峰在1500~1300 cm-1区域为下凹曲线,因此根据上述规则计算出的N-H 弯曲振动峰的面积和高度均为负值。无论是用峰面积还是峰高度作为量化参数,白矾与铵明矾的N-H 弯曲振动峰强度都有极显著差异(P<0.001)。也就是说,1436 cm-1附近的NH 弯曲振动峰可以作为鉴别白矾与铵明矾的专属性特征峰。
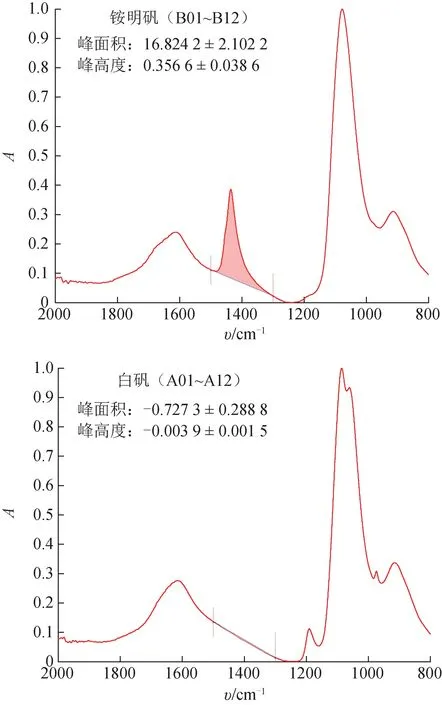
图5 市售真、伪白矾样品的MIR光谱中铵离子特征峰强度
3.3 白矾与铵明矾混合物的MIR光谱检测
白矾(A01)与铵明矾(B01)不同比例混合物的MIR 光谱见图6。可以看到,随着混合物中铵明矾含量增加,1436 cm-1附近的N-H 弯曲振动峰也逐渐增强。无论是用峰面积还是峰高度评价,白矾(A01)与铵明矾(B01)混合物MIR 光谱中N-H 弯曲振动峰强度与铵明矾含量都有良好的线性关系(图7)。但是,不同白矾与铵明矾的混合物MIR 光谱放在一起时,N-H 弯曲振动峰强度与铵明矾含量的线性度较差(图8)。主要原因可能是水分和杂质含量比例不同,导致不同铵明矾样品的N-H 弯曲振动峰的强度不同(图4)。因此,MIR 光谱中N-H 弯曲振动峰暂时不宜作为白矾样品中掺入铵明矾的定量检测,应当作为白矾样品掺假的定性或限量检测方法。

图6 白矾(A01)与铵明矾(B01)不同比例混合物的MIR光谱中铵离子特征峰强度
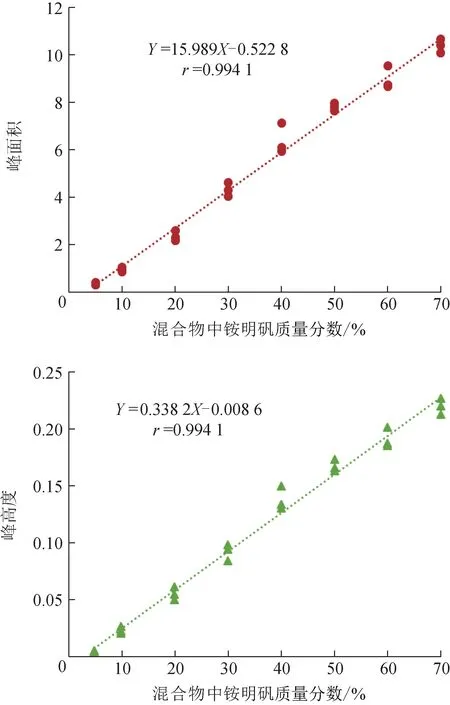
图7 白矾(A01)与铵明矾(B01)混合物MIR光谱中铵离子特征峰强度与铵明矾质量分数的线性关系

图8 不同白矾与铵明矾混合物的MIR光谱中铵离子特征峰强度
如图5 所示,11 批白矾样品中N-H 弯曲振动峰的峰面积均值为-0.727 3 cm-1,标准差为0.288 8 cm-1,99%置信区间上限为0.017 7 cm-1;峰高度均值为-0.003 9,标准差为0.001 5,99%置信区间上限为-0.000 1。也就是说,以1500、1300 cm-12 点建立局部基线,如果待测白矾样品的1500~1300 cm-1区域光谱积分面积超过0.017 7 cm-1,或者1450~1430 cm-1区域光谱最大高度超过-0.000 1,可以判断该样品为铵明矾或者掺入铵明矾的白矾。
11 批白矾样品的MIR 光谱中N-H 弯曲振动峰的峰面积最高值为-0.240 0 cm-1,峰高度最高值为-0.002 3 A,按照上述规则均可判断为未掺假样品;12 批次铵明矾样品的N-H 弯曲振动峰的峰面积最低值为14.220 0 cm-1,峰高度最低值为0.308 5,按照上述规则均可判断为掺假样品。
本研究进一步用6 个白矾样品、6 个铵明矾样品分别配对制成铵明矾质量分数为1%、5%、10%、20%的混合物,每个样品测试3 次,根据上述N-H弯曲振动峰识别规则判断混合物是否为掺假样品。如图8所示,铵明矾质量分数为10%的6组混合物样品、铵明矾质量分数为20%的6组混合物样品的各3次检测结果均为掺假样品,而铵明矾质量分数为1%的1组混合物样品(A01+B01)的3次检测结果均为假阴性。铵明矾质量分数为5%的5 组混合物样品的各3 次检测结果均为掺假样品,只有1 组混合物(A05+B10)的1 次检测结果为假阴性、2 次检测结果为掺假样品,可能是该样品不均匀所致。综上,可以认为铵明矾质量分数不低于5%的掺假白矾样品基本都能被准确识别。十二水合硫酸铝铵相对分子质量为453.33,氮的相对原子质量是14.01,由此推算铵明矾中氮元素质量分数约为3%。白矾中含有5%铵明矾时,氮元素质量分数约为0.15%。《中国药典》2020 年版白矾的铵盐检查项规定样品中含铵盐以总氮计不超过0.3%。也就是说,MIR光谱对于白矾中铵盐的检出限完全可以满足药典要求。
3.4 MIR光谱不同测试方法比较
溴化钾(KBr)压片法与ATR 法是目前固体样品MIR 光谱测试最常用的方法。白矾、铵明矾的主要成分是含有结晶水的离子化合物,使用压片法时样品可能与作为稀释剂的KBr 产生一定程度的离子相互作用,加之实验室湿度、样品粒度、样品与稀释剂混匀程度等因素,都可能影响样品谱图形状。而且压片法需要消耗KBr,使用研磨和压片工具,也增加了检测过程的时间和物质成本。使用ATR 法测试MIR 光谱时,样品直接加载于内反射晶体表面即可,操作过程更加简便、快速,可能影响样品谱图形状的干扰因素也更少。因此,本研究选择ATR法测试白矾与铵明矾样品的MIR 光谱。本研究过程中也比较了ATR 法与KBr 压片法测试的白矾与铵明矾样品的MIR 光谱,2 种方法获得的光谱特征基本一致,主要差异在于KBr 压片法测得的吸收峰位置波数普遍高于ATR 法(图9)。例如,ATR 法测量的MIR光谱中铵明矾的N-H 弯曲振动峰在1436 cm-1附近,而KBr 压片法测量的该峰位置在1441 cm-1附近。因此,将N-H 弯曲振动峰的出现位置放宽到1450~1430 cm-1区域是更加合理、耐用的规则。
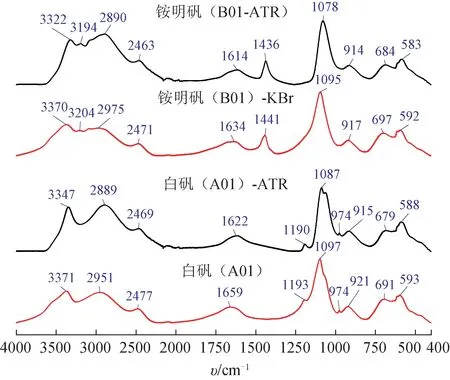
图9 同一仪器不同方法测试的白矾与铵明矾的MIR光谱
4 结论
综上所述,MIR 光谱中1450~1430 cm-1区域的铵离子N-H 弯曲振动峰可作为专属特征峰,用于鉴别白矾与铵明矾,或者识别掺入铵明矾(不低于5%)的白矾。该方法适合于不同实验室、不同品牌型号的MIR 光谱仪,可选择ATR 法或KBr 压片法测试样品MIR 光谱。考虑到可能存在的其他伪品或掺假物质,可以将白矾样品与十二水合硫酸铝钾的MIR 整体特征一致作为基本要求,同时将N-H 弯曲振动峰作为靶向检查铵明矾或铵离子的指标。因此,建议将白矾的MIR 光谱鉴别标准设定为“取本品粉末适量,衰减全反射法或溴化钾压片法制备供试品,照红外分光光度法(药典通则0402)试验,供试品红外吸收图谱应与十二水合硫酸铝钾[KAl(SO4)2·12H2O] 具有相同的特征吸收峰,且在1450~1430 cm-1区域内不得出现吸收峰”。
[利益冲突]本文不存在任何利益冲突。
