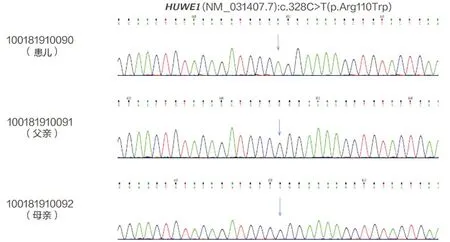
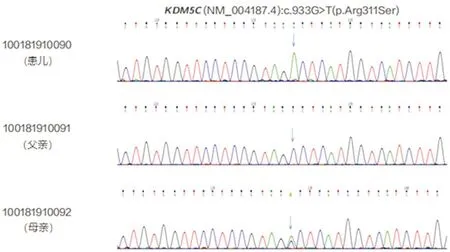
HUWE1基因突变合并KDM5C基因突变导致X连锁的智力障碍1例
2024-04-25刘青陈波刘畅
刘青,陈波,刘畅
患儿,男,13岁,因“生长迟缓10余年”于2022年4月21日入院。患儿出生史无特殊,生长发育史落后,6个月时诊断为“囟门早闭、颅缝早闭”。患儿目前为六年级,言语欠清晰,能正确对答,学习成绩较差,好多动,注意力不集中,脾气暴躁,易怒,有自残和攻击行为。患儿父母、姐姐及家族中无类似疾病患者。体格检查:身高143 cm( 图1 患儿面容、口腔、胸廓和手指表现 图2 患儿头颅MR影像学表现 中国修订韦氏儿童智力量表(C-WISC)结果提示,患儿言语、操作和全量表智商分别为 63、70和 63,其百分位分别为0.80、2.60、0.80。Conners父母问卷:学习问题表现为做事有始无终,注意力不能集中,在学习过程中不能持之以恒,学习成绩下降。基因检测:检出与受检者临床信息相关的1个新发致病性变异为HUWE1(NM_031407.7):c.328 C> T(p.Arg110Trp)(图3),参考美国医学遗传学与基因组学学会(ACMG)和美国分子病理学会(AMP)对于遗传变异的分类标准和指南[1],该变异为致病性变异(PM6_Strong+PM2+PM5+PS4_Moderate);检出与受检者临床信息可能相关的1个母源性临床意义未明变异为KDM5C(NM_004187.4):c.933 G> T(p.Arg311Ser),exon7(图4),该变异证据链PM2,综合判定为临床意义未明变异(VUS)。结合临床表型及基因检测结果,诊断为HUWE1基因突变合并KDM5C基因突变导致X连锁的智力障碍。给予对症治疗及功能康复训练,随访12个月,目前患儿身高147 cm,吐字较前清晰,言语流利程度稍有进步,运动较前稍有进步,专注力较前集中,能单独看书、听课10余分钟,学习数学等科目仍有困难,攻击性行为未见好转。 图3 患儿及其父母HUWE1基因测序图 图4 患儿及其父母KDM5C基因测序图 讨 论智力障碍(intellectual disability,ID)是一种复杂的神经发育状况,发病率为1%~3%,而在X染色体上的基因发生致病性突变所引起的先天性精神发育迟滞,称为X连锁的智力障碍(X-linked intellectual disability,XLID)。目前已在X染色体上鉴定出120个基因与XLID的发生有关[2],约占ID患者的10%以上[3]。 HUWE1基因定位于Xp11.22上,在神经元发育、增殖过程中广泛表达,可促进突触的发生,并有助于维持基因组完整性[4]。HUWE1基因突变诱发HUWE1功能增加(又称功能获得)或功能丧失减少从而导致XLID 的发生[5]。通过检索及多番对比国内外文献后,本研究发现 HUWE1突变的患者通常表现为:中度至重度的智力发育落后、身材矮小、手脚较小、严重的言语发育落后和非特异性的面部特征异常(如深陷的眼睛、宽大的鼻尖等)[5]。 本例患儿除以上临床体征外,还有颅缝早闭(6个月之前颅缝早闭、小头颅)及Chiari畸形(头颅MR:小脑扁桃体下疝畸形)等新的临床特点。通过文献复习发现HUWE1(c.328 C> T;p.Arg110Trp)同一位点突变的2例男性患儿也具有此特点,但其中1例患儿还具有脊柱侧凸、小脚趾并指的附加特征[6-7]。这提示即使均为HUWE1基因突变甚至突变位点相同所造成的临床表型也会有细微差别。同时由于HUWE1基因突变病例数极为罕见,暂无法识别HUWE1变异的个体与该基因相关的临床表型,需进一步阐明基因型—临床表型的相关性。 本例患儿同时伴随牙列稀疏、易怒、自伤行为的特点,与目前已报道的HUWE1基因突变临床表型并不完全一致,查阅基因报告发现患儿还存在1个可能相关的母源性临床意义未明变异:KDM5C基因(c.933 G> T;p.Arg311Ser)突变,该基因突变可导致一种罕见的呈 X 染色体连锁隐性遗传方式发病的先天性智力障碍疾病——Claes-Jensen综合征(mental retardation, X-linked, syndromic,Claes-Jensen type, MRXSCJ),表现为轻至重度的精神发育迟滞,占男性XLID患者的3%[8-10]。检索国内文献已证实致病性 KDM5C 变异的男性都具有颅面异常、癫痫和身材矮小的特征,并且拥有牙列稀疏、易怒、自伤行为的特点,本例患儿缺牙症、智力低下明显伴有多动障碍、注意力不集中,学习困难、易怒等临床特征均符合KDM5C基因突变的临床表型。 结合患儿基因检测结果及临床症状,新发致病性变异 HUWE1(c.328 C>T;p.Arg110Trp)与患儿临床症状相符,提示该基因变异是患儿的患病原因,但新发变异检出并不能排除父母中某一方为嵌合体的情况。建议患儿父母再生育时考虑通过产前诊断或胚胎植入前遗传学诊断(PGD)技术避免同样病症患儿的出生。本例患儿同时还发现另外1个VUS 变异 KDM5C(c.933 G>T;p.Arg311Ser),其是否致病(单独或联合致病变异而使病情加重)则需要进一步分析临床随访和家系基因检测结果,受检者的兄弟姐妹及其他有相似临床症状的直系亲属同时检测该变异,有助进一步分析此基因变异与疾病的相关性。 迄今为止,对于XLID患儿无特效治疗方法,以对症治疗、康复功能训练治疗为主。对于身高偏矮者可考虑使用重组人生长激素治疗,可促进身高增长[11]。本例患儿家长无促生长治疗意愿,故现给予康复对症治疗。 通过报道此例患儿,从遗传学角度明确了其病因,扩大了HUWE1、KDM5C基因的变异谱。但仍需要继续收集其相关资料,加强对此病的认识,以便将来更快识别HUWE1基因突变及KDM5C基因突变的个体,明确与该基因相关的表型,帮助临床工作中对于此类患儿的早发现及精准诊断,减少临床误诊和漏诊,以及为家系的产前诊断提供参考。