Graphene–calcium carbonate coating to improve the degradation resistance and mechanical integrity of a biodegradable implant
2024-04-18LokshChouharyParamaChakrabortyBanrjSinghRamanDrrkLoboChristophrEastonMainakMajumrFrankWittrglr
Loksh Chouhary ,Parama Chakraborty Banrj ,R.K.Singh Raman ,Drrk E.Lobo ,Christophr D.Easton ,Mainak Majumr ,Frank Witt,Jörg F.Lölr
aDepartment of Mechanical and Aerospace Engineering, Monash University, Clayton Campus, Melbourne, VIC 3800, Australia
b Department of Chemical and Biological Engineering, Monash University, Clayton Campus, Melbourne, VIC 3800, Australia
c Nanoscale Science and Engineering Laboratory (NSEL), Bldg 37, Monash University, Clayton Campus, Melbourne, VIC 3800, Australia
d CSIRO Materials Science and Engineering, Bayview Avenue, Clayton, VIC 3168, Australia
e Julius Wolff Institute and Berlin Brandenburg Center for Regenerative Therapies, Charité– Universitätsmedizin Berlin, Augustenburger Platz 1, 13353 Berlin, Germany
fLaboratory of Metal Physics and Technology, Department of Materials, ETH Zurich, 8093 Zurich, Switzerland
Abstract Biodegradable implants are critical for regenerative orthopaedic procedures,but they may suffer from too fast corrosion in human-body environment.This necessitates the synthesis of a suitable coating that may improve the corrosion resistance of these implants without compromising their mechanical integrity.In this study,an AZ91 magnesium alloy,as a representative for a biodegradable Mg implant material,was modified with a thin reduced graphene oxide (RGO)-calcium carbonate (CaCO3) composite coating.Detailed analytical and in-vitro electrochemical characterization reveals that this coating significantly improves the corrosion resistance and mechanical integrity,and thus has the potential to greatly extend the related application field.
Keywords: Graphene coating;Biodegradable implant;Hydroxyapatite;Corrosion;Magnesium alloy.
1.Introduction
Traditional implant materials including stainless steels,titanium alloys and cobalt–chromium alloys possess good resistance to corrosion,wear and fatigue along with excellent load-bearing capabilities.However,it is well known that these alloys contribute to various negative effects including stress shielding [1]and inflammation of local tissues due to potential release of cytotoxic ions [2],and in many cases an implant removal surgery is necessary.Thus,in biomedical applications,temporary implants that ‘biocorrode’ are currently attracting significant interest [3–8].Polymers and metals are two material classes that have been considered widely for biodegradable implant applications due to their inherent ability to corrode away within the human body,which abrogates the second surgery for implant removal [9–11];however the corrosion/degradation products should be harmless.Among them,metallic materials such as magnesium alloys are of greater interest due to their advantageous mechanical properties.However,rapid corrosion of magnesium-based implants in body fluid is a major bottleneck with respect to their wider use [1,12,13].This necessitates the development of bioactive surface treatments to control the corrosion rate.
In this context,biocompatibility,osteoconduction and osteogeneration capability of calcium phosphate(Ca–P)coatings(such as hydroxyapatite,brushite and octacalcium phosphate)make them attractive for enhancing the corrosion resistance of biodegradable magnesium implants [14–20].Various approaches,including thermal spraying,sol-gel processing,biomimetic coatings,electrochemical and electrophoretic depositions,spray coating,physical vapor deposition,magnetron sputtering and pulsed laser deposition,have been employed to develop Ca–P coatings on magnesium-based implants [18–22].It is to be noted that direct deposition of hydroxyapatite on a magnesium alloy in the presence of simulated body fluid (SBF) is generally difficult [19].In general,the majority of the Ca–P coatings synthesized on magnesium alloys can easily develop cracks,which can significantly compromise the corrosion barrier properties due to the coatings [20].
Graphene,a sp2-bonded carbon layer of graphite with a thickness of a single atom,has triggered widespread research interest,resulting from its excellent thermal,mechanical and electrical properties [23,24].Besides these attributes,graphene possesses remarkable chemical inertness [25],impermeability,and hydrophobicity [26,27],which are ideal for corrosion barrier coatings [28].Recently,a composite consisting of bone bioactive minerals (such as calcium carbonate and calcium phosphate) and graphene has been realized with great potential for biomedical applications [19].Furthermore,graphene and graphene oxide (GO) have been reported to facilitate adhesion and proliferation of SAOS-2 cells (human osteoblast-like cell line),and have the potential to induce the differentiation of mesenchymal stromal cells into the osteoblast lineage [29–31].Kim et al.[32]synthesized 150 μm thick free-standing graphene/calcium carbonate(CaCO3)films,which,after incubation in simulated body fluid(SBF) for 4 days,transformed into graphene/hydroxyapatite(Hap)composites.These free-standing graphene/Hap composite films showed high viability of mouse osteoblast (MC3T3-E1) cells.Lahiri et al.[33]reported low concentrations of graphene fillers to provide an improved biocompatibility to bone cells.These studies all confirmed the biocompatibility of graphene,GO and CaCO3in individual form and as composites.
Recently,a few reports have emerged on the corrosion barrier properties of GO/Hap coatings developed on magnesium and titanium implants [18-20,34].Li et al.[18]reported cathodic electrophoretic deposition of a 35 μm thick GO/Hap coating on a titanium substrate,which improved the corrosion resistance of the metallic substrate by an order of magnitude.Gao et al.[19]reported an order of magnitude improvement in corrosion resistance from a 34 μm thick GO/Hap coating on an AZ91 magnesium alloy.However,the time-consuming synthesis process of 2 days to grow the optimized coating limits the practical use of this particular coating.A thin GO/HapNP(hydroxyapatite nanoparticle) coating with 1 μm thickness was developed by Santos et al.[20],but this coating provided little protection to the high-purity magnesium substrate.It is well known that inhibition of implant corrosion during the first few days after surgery is critical,as during this period the risk of inflammation is highest.However,most of the reports so far only mention the coatings’ corrosion barrier properties soon after their immersion in body fluid (the longest immersion time so far has only been 24 h)[19].There are also no detailed reports on the coatings’ mechanical integrity under load-bearing conditions after their exposure to the body fluid.
In the present study,we have developed a thin graphene oxide (RGO)/CaCO3composite coating on an AZ91 magnesium alloy using a facile and faster (2 h) synthesis process,which promoted enhanced hydroxyapatite formation on the coated metallic substrate in the presence of modified simulated body fluid (m-SBF).Magnesium alloys in general reveal good biocompatibility,non-toxic degradation products,and superior mechanical compatibility with bone (as compared to the traditional alloys used as implants) [4,9,35-39].The present study reports the corrosion resistance of(RGO)/CaCO3composite coating during the first five days with an elaborated insight into the degradation mechanisms.The synergistic interaction between RGO and CaCO3significantly enhances the corrosion resistance and the mechanical integrity of the metallic substrate in presence ofm-SBF.
2.Material and methods
2.1. Material used
Rectangular AZ91D magnesium alloy specimens (Al:8.8 wt.%,Zn: 0.8 wt.%,Mn: 0.2 wt.%,Mg: balance) of 1.3 cm×1.3 cm×0.9 cm dimensions were machined from their as-cast ingots.These specimens were progressively ground with silicon carbide papers down to 2500 grit,thoroughly rinsed with acetone and deionized water,and subsequently dried with compressed air.
2.2. Coating method
The details of graphene oxide (GO) synthesis is explained in Refs.[40,41].GO (∼0.16 wt.%) was dispersed in deionized water and then subjected to ultrasonication for 1 h.The resultant dispersion was centrifuged at 3000 rpm for 30 min.Concurrently,0.3 M calcium chloride dihydrate(CaCl2∙2H2O) solution (180 ml) was mixed with 0.6 M ammonium hydroxide (NH4OH) solution (120 ml).The GO dispersion (180 ml) was added to this solution mixture followed by a vigorous stirring.The pH of this coating solution was 11.7.The rectangular specimens of the AZ91D alloy (5 specimens at a time) were then immersed in the coating solution using nylon strings for 60 min at the ambient temperature.During the immersion,carbon dioxide (CO2) gas was purged at ∼1 l/min to facilitate the formation of GO–CaCO3coating on AZ91D,and the solution was stirred at 120 rpm.The suspended AZ91D samples were coated on both sides.
In order to convert GO–CaCO3into RGO–CaCO3,the GO–CaCO3coated specimens were subsequently immersed in a reducing mixture of 0.15 ml of hydrazine(35 wt.%),1 ml of NH4OH (0.28 wt.%) and 300 ml of water,and then cured at 95 °C for 60 min.To understand the barrier properties of the CaCO3coating without RGO,a few of the AZ91 specimens were coated following the same process as the GO–CaCO3coating,where CO2gas was purged for 1 h into the solution mixture of CaCl2and NH4OH without any addition of GO dispersion.The CaCO3coated specimens were then dried in an oven at 95 °C for 60 min.
2.3. Scanning electron microscopy (SEM)
A JEOL 7001F scanning electron microscope (SEM) that operated at 15 kV was used to determine the coating morphology.The cross-sections were obtained using a FEI Helios Nanolab 600 focused ion beam (FIB)-SEM,operating at a vacuum level of below 1×10-3Pa.Ga ions were used to mill the sample at an accelerating voltage of 30 kV with a 93 pA current,while the images were taken by the SEM operating at 5 kV and at a tilt angle of 52° All samples were coated with 1 nm thick conductive carbon coating to avoid charging.
2.4. X-ray diffraction (XRD) analysis
The chemical constituents of the coating were analysed by X-ray diffraction (XRD) using Cu Kαradiation generated at 40 kV and 25 mA.The scan rate was 2° per minute and the step size was 0.02°.
2.5. Raman spectroscopy
A RenishawR○Confocal micro-Raman Spectrometer was used to obtain the Raman spectra of the specimens.The spectrometer was equipped with a helium-neon laser,operating at 10% power.The laser spot size was 1 μm.The scans(10 s) were performed between wave numbers of 100 cm-1and 3200 cm-1[42].
2.6. Attenuated total reflectance-Fourier transformed infrared spectroscopy (ATR-FTIR)
A PerkinElmerR○Spectrum 100 series FTIR spectrometer was used to collect ATR-FTIR spectra for the coated and uncoated specimens.The spectra were collected in the mid-IR range(500-4000 cm-1),and at least 128 scans were obtained for each specimen.The spectral resolution was 8 cm-1.
2.7. X-ray photoelectron spectroscopy (XPS)
An AXIS Ultra DLD spectrometer was used to perform XPS analysis.The spectrometer consisted of an Al Kαsource operating at a power of 90 W.It also had a hemispherical analyser operating in a fixed transmission mode and an aperture with an analysis area of 0.3 mm×0.7 mm.Typical pressure in the vacuum chamber was ∼10-8mbar,whereas a pass energy of 160 eV was used to acquire the spectra.
CasaXPS processing software,version 2.3.15,was deployed to analyse the survey spectra,and integral peak intensities and sensitivity factors were used to calculate the atomic concentrations [42].The C 1 s peak (285 eV) was considered to correct the binding-energy scale for sample charging [42].
2.8. Electrochemical testing
The electrochemical experiments were performed at 37 °C inm-SBF solution,which was prepared following the established procedure by Oyane et al.[43].The suitability ofm-SBF as an in-vitro electrolyte has been discussed in detail in the supporting information (section S1).
A water bath and a submersible pump were used to circulatem-SBF through the electrochemical testing cell.A simple graphic of this cell is shown in Figure S1(supporting information,section S2).The circulation ofm-SBF also helped avoiding localized changes in pH of the test solution throughout the experiment.The electrochemical experiments were performed using a PARSTATR○2273 potentiostat and a three-electrode cell: the coated and uncoated magnesium alloy specimens with an exposed area of ∼0.8 cm2were the working electrode;a saturated calomel electrode (SCE) was the reference electrode;and platinum was used as the counter electrode.
Open circuit potential (OCP) was measured to ensure electrochemical stability [44,45].The potentiodynamic polarisation experiments were performed in them-SBF with a potential sweep rate of 0.5 mV/s.Electrochemical impedance spectroscopy (EIS) experiments were performed by applying a sinusoidal potential amplitude of 10 mV at OCP.Impedance response was measured over the frequency range of 1 MHz to 10 mHz.It is well known that artefacts can be present in the high-frequency range and significant scatter can be present in the low-frequency impedance data (especially for fast corroding metallic systems).The data analysis was therefore only performed within the frequency range of 100 kHz to 50 mHz,using the PAR ZSimpWin software package.The electrochemical measurements were repeated at least three times to examine reproducibility.
3.Results and discussion
3.1. Morphological and chemical characterization
Fig.1a shows the development of a uniform and dense RGO-CaCO3coating on the AZ91 substrate.A highmagnification micrograph (Fig.1b) shows that the coating primarily consisted of spherical vaterite crystals,which were wrapped by interconnected thin membrane-like RGO films.In contrast,the CaCO3coating without RGO had a predominance of rhombohedral CaCO3crystals with occasional presence of vaterite spheres (Fig.1c and d).The cross-sectional analysis of the coated specimens reveals that a uniform and thin (∼0.5 μm) RGO-CaCO3coating (Fig.1e) developed on the magnesium alloy,whereas the CaCO3coating without RGO (Fig.1f) was apparently porous and thicker (∼2.5 μm).In this context it is important to note that the coating thicknesses mentioned are the composite thicknesses of the metal oxide/hydroxide layers and the coatings.

Fig.1.SEM micrographs of the surface morphology of (a) and (b) RGO-CaCO3 coated,and,(c) and (d) CaCO3 coated magnesium alloy.Cross-sections of(e) RGO-CaCO3 coated and (f) CaCO3 coated specimens.
In order to verify that the spherical crystals and the thin membrane-like structures were indeed of vaterite and RGO,respectively,XRD and Raman spectra were performed on the RGO-CaCO3coated,only CaCO3coated and uncoated specimens.The XRD peaks (Fig.2a) at 32.7°,41.7° and 43.8° indicate the predominance of vaterite crystals (PDF card # 033–0268) in the RGO-CaCO3coated specimen as opposed to the calcite peaks (at 29.4° and 48.5°,PDF card #047–1743) that were prevalent in the case of the only CaCO3coating.XRD spectra of the RGO-CaCO3coating did not show any peak related to RGO,which may be due to the thin membrane-like structure of RGO in this composite coating.However,Raman spectra confirmed the presence of RGO(Fig.2b),which are characterized by the D peak (∼1350 cm-1) assigned to the breathing mode of к-point phonons of A1gsymmetry,and the G peak (∼1590 cm-1) corresponding to the E2gphonon of the sp2hybridization [28,40,41,46,47].

Fig.2.(a) XRD,(b) Raman,and (c) ATR-FTIR spectra of the RGO-CaCO3 coated,CaCO3 coated,and uncoated specimens.
CaCO3is also Raman active [48,49]with characteristic peaks appearing at 280,714,1085 and 1435 cm-1.The most intense peak at 1085 cm-1arises due to the symmetric stretching (ν1vibration) of carbonate.The 280 cm-1peak arises due to lattice modes and the 575 cm-1peak arises due to the in-plane bending (ν4vibration) of carbonate.A very weak peak at around 1435 cm-1arises due to the antisymmetric stretch (ν3vibration) of carbonate [48,49].The characteristic peaks of both RGO and CaCO3were present in the Raman spectra of the RGO-CaCO3coating,which further confirms the formation of a composite between RGO and CaCO3.Fig.2b shows that RGO is more Raman active than CaCO3,which can be attributed to the large number of aromatic rings in its structure.Consistent with the observation of thin membrane-like RGO films in the RGO-CaCO3coating(Fig.1b),the Raman spectra of this coating shows presence of a broad 2D peak at ∼2670 cm-1.The 2D to G ratio is 0.2 ± 0.02,which corresponds to 3–4 layers of RGO [50].
The FTIR spectrum (Fig.2c) of the RGO-CaCO3coated specimen consists of the characteristic peaks of CaCO3(883 cm-1,corresponding to theν2out-of-plane bend,and a broad peak in the range of 1400–1500 cm-1,corresponding to theν3asymmetric stretch) [51,52],along with a peak at 1525 cm-1,corresponding to the C=C bond of RGO.In summary,the SEM micrographs,XRD data,Raman spectra and FTIR analysis confirm the formation of an RGO-CaCO3composite and the presence of vaterite and RGO in the RGO–CaCO3coating.
GO possesses several oxygen-containing groups,such as hydroxyl,epoxy,carboxyl and other carbonyl groups [53–55],most of which are deoxygenated by hydrazine reduction [56].However,the reduction of carboxyl groups is unlikely via hydrazine under the given conditions [56,57].These negatively charged carboxyl groups present in RGO can associate with the Ca2+of vaterite by electrostatic interaction and hinder the dissolution of metastable vaterite [58].In the absence of such hindrance,the vaterite crystals may have dissolved and recrystallised as more stable allotrope,i.e.as calcite (having rhombohedral crystals),as seen in the case of only CaCO3coating without RGO (Fig.1c and d).
3.2. Electrochemical characterisation
Fig.3a shows the potentiodynamic polarisation plots of the RGO-CaCO3coated,only CaCO3coated and uncoated magnesium alloy at 2 h of immersion inm-SBF.The anodic and the cathodic current densities of the RGO-CaCO3coated specimen were an order of magnitude smaller than those of the only CaCO3coated and the uncoated alloy.In order to investigate the time-dependent barrier properties of the RGO-CaCO3coating,the coated specimens were pre-immersed for different durations (2 h,25 h and 120 h) inm-SBF,before the potentiodynamic polarisation tests.Fig.3b shows that the corrosion potential (Ecorr) of the RGO-CaCO3coated specimen became more positive (at 25 h,100 mV more positive and at 120 h,200 mV more positive than that after 2 h) with increasing immersion time inm-SBF,suggesting a decrease in the coated alloy’s corrosion susceptibility with increasing immersion time [45].Up to an overpotential of -1.35 V vs SCE,the anodic current density of the RGO-CaCO3coated specimen immersed for 25 h inm-SBF was an order of magnitude lower than that at 2 h of immersion.Although at 120 h of pre-immersion,the anodic current density increased as compared to that at 25 h,it still remained similar to that obtained at 2 h of pre-immersion inm-SBF.This is in agreement with the EIS data (Fig.3c),where the magnitude of impedance(|Z|) at the lowest frequency,which is a measure of corrosion resistance [59],increased in the initial 25 h of immersion inm-SBF for the coated samples and then gradually decreased over time.Corresponding Bode modulus plots are provided in the supporting information,Figures S2-S4.However,at 120 h of immersion,the magnitude of impedance of the RGO–CaCO3coated specimen was an order of magnitude higher than that of the CaCO3coated and the uncoated alloy.This can be attributed to the highly porous nature of the CaCO3coating,which consists of loosely scattered rhombohedral calcite crystals (Fig.1c,d and f) and thus provides little protection to the magnesium alloy surface (Fig.3a and c).
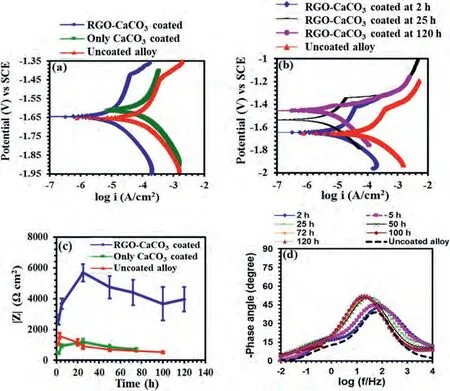
Fig.3.(a) Potentiodynamic polarisation of the RGO-CaCO3 coated,CaCO3 coated,and uncoated alloy after 2 h of immersion in m-SBF.(b) Potentiodynamic polarization of the RGO-CaCO3 coated specimen after pre-immersion in m-SBF for different durations (2 h,25 h and 120 h).The polarization plot of the uncoated alloy was recorded after 2 h of immersion in m-SBF.(c) Magnitude of impedance at the lowest frequency (10 mHz) of the RGO-CaCO3 coated,CaCO3 coated,and uncoated specimens at different durations of immersion in m-SBF,and (d) Bode phase-angle plots of the RGO-CaCO3 coated specimen at different durations of immersion in m-SBF.The Bode plot of the uncoated specimen was generated after 2 h of immersion in m-SBF.
The phase angles (Fig.3d) of the RGO-CaCO3coated and uncoated specimens were in the range of -45 to -50°,suggesting predominance of diffusion-controlled processes at the metal/electrolyte interfaces [60].During the first 5 h of immersion,the RGO-CaCO3coated specimen showed a broader time constant at the same frequency range than that of the uncoated alloy,which can be attributed to the overlap of the time constants arising due to the responses from the RGO-CaCO3coating/electrolyte and the metal/electrolyte interfaces [61].However,with increasing immersion times,the time constants shifted towards lower frequencies,which may be attributed to a gradual degradation of the coating,generating an increased rate of charge transfer at the metal/electrolyte interface.This aspect will be illustrated in detail in the following section.
3.3. Mechanistic insight
The electrochemical results raised the following questions:(i) Why does the corrosion resistance of the RGO-CaCO3coating increases during the initial 25 h of immersion inm-SBF? (ii) Why is the corrosion resistance of the RGOCaCO3coated specimen higher than that of the CaCO3coated specimen? (iii) What are the electrochemical processes at the RGO-CaCO3coating/electrolyte and metal/electrolyte interfaces? And how did these processes and their kinetics influence the barrier properties of the RGO-CaCO3coating at different immersion durations inm-SBF? In order to answer these questions,a two-pronged strategy was employed: (i)SEM and XPS were performed to monitor the physical and chemical changes of the RGO-CaCO3coating inm-SBF at different immersion durations,and (ii) an electrical equivalent circuit (EEC) was used to analyse the EIS data to obtain mechanistic insight into the electrochemistry at the different interfaces.
Fig.4 shows that the RGO-CaCO3coated specimens after immersion inm-SBF for 25 h (Fig.4a) and 120 h(Fig.4b) were covered with spheres of lathlike crystals,whereas no such formation was observed in the case of the CaCO3coated sample (Fig.4c).As the solubility constant,Ks,of vaterite (logKs=-7.91,at 25 °C) suggests,it is highly soluble (CaCO3→Ca2++CO32-) in aqueous solutions[58].Thus,the formation of the lathlike structures on the RGO–CaCO3coated specimens may be a result of the partial dissolution of spherical vaterite in the presence ofm-SBF.In fact,it is well known [32,62]that Ca2+ions produced upon dissolution of vaterite can combine with PO43-available inm-SBF and nucleate lathlike hydroxyapatite (Hap) crystals on the dissolving spherical vaterite surface (Fig.4a and 4b).

Fig.4.SEM micrographs of an RGO-CaCO3 coated specimen immersed in m-SBF for (a) 25 h and (b) 120 h.(c) SEM micrograph of the surface morphology of a CaCO3 coated specimen at 72 h of immersion in m-SBF.
To confirm the formation of hydroxyapatite on RGOCaCO3coated specimens after immersion inm-SBF,X-ray photoelectron spectroscopy (XPS) was undertaken.Specifically,a pristine RGO-CaCO3coated sample and an RGOCaCO3coated sample immersed inm-SBF for 120 h were analysed to confirm the presence of phosphorous (P),an element unique to hydroxyapatite in this particular case.Fig.5 shows high-resolution spectra of the P 2p region for the two samples.As expected,no P was detected on the pristine sample,while P was clearly present on the RGO-CaCO3coated specimen immersed inm-SBF.Quantification of the surface spectra resulted in an atomic concentration of 0.7% P on the RGO-CaCO3sample immersed inm-SBF.
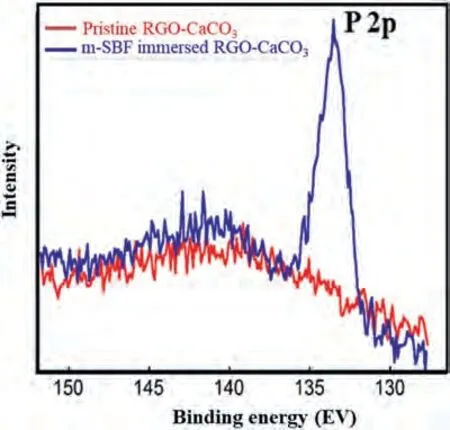
Fig.5.Presence of the P 2p peak in the XPS scan of the immersed RGOCaCO3 specimen indicates formation of Hap on the coated specimen surface due to immersion in m-SBF for 120 h.
K2HPO4·3H2O is the only reagent inm-SBF that contains P.Thus,it is possible to confirm indirectly that PO43-has coupled with Ca2+to form hydroxyapatite,rather than to simply physically adsorb in the form of K2HPO4,by examining the surface for the presence of K.This test assumes that in the case of hydroxyapatite formation,K is removed from the surface after immersion inm-SBF.For small amounts of K,it may be difficult to confirm the presence of the K 2p peak from low-resolution survey spectra due to its proximity to the C 1 s peak.In addition,the large amount of Mg present results in a set of Auger peaks with large intensity that occur in the region of K 2 s.Because of these complications,it was not possible to ascertain the absence of K from the survey data alone.As a result,the only reliable way to confirm the presence of a small amount of K on the surface was to examine the high-resolution C 1 s spectra with a sufficiently large binding-energy window to capture the K 2p doublet.As observed in Figure S6(supporting information section S7),there is no intensity present above the background signal in the region for K 2p,as defined by the highlighted region.All elements present were identified from the survey spectra.Table S2 (supporting information section S7) reveals the elemental quantification derived from the survey spectra in atomic%and atomic ratios (X/C) for the samples RGO-CaCO3andm-SBF immersed RGO-CaCO3.There is no entry for K in Table S2 as no signal from K 2p or K 2 s was observed.Therefore,the presence of P on the surface and the absence of the K 2p peak illustrate that P is likely present on the surface in the form of hydroxyapatite,considering also the other elements present in Table S2.More detailed discussion on the XPS analysis is provided in the supporting information section S7.
Fig.4a suggests that at 25 h of immersion the spherical vaterite crystals retained their shape and did not completely dissolve although they were covered with Hap crystals.It is to be noted here that Hap gradually dissolves in SBF [63],and hence,when deposited on a metallic substrate,it can delay the corrosion of a metal substrate but cannot completely stop it [37,64,65].This is one of the primary advantages of using Hap coatings for biodegradable implants.Since the corrosion resistance (Figs.3b and 3c) of the RGO-CaCO3coated specimen increased during immersion until 25 h,it is reasonable to assume that during this initial stage of immersion,the formation rate of Hap was higher than the dissolution rate.Interestingly,even though the electrochemical data(Figs.3b and 3c) showed a gradual decline in the corrosion resistance of the RGO-CaCO3coating,no significant morphological change was observed in the specimen after 120 h of immersion inm-SBF (Fig.4b).This disparity between the electrochemical data and SEM images can be explained if one considers the limitations of the microscopic techniques in providing any information on the rate of morphological changes related to a slow dynamic physical process.
In order to obtain mechanistic insight into the influence of the formation and dissolution rate of Hap on the electrochemical kinetics at the different interfaces (RGO-CaCO3coating/electrolyte and metal/electrolyte),and consequently,on the corrosion resistance of the RGO-CaCO3coated specimens at different immersion durations,an electrical equivalent circuit (EEC),Rs(Qg[Rg(QdlRc)]),was employed to analyse the EIS data.This EEC (Fig.6a) has been extensively used in the literature [28,45,61,66-73].In the present study,complex nonlinear least squares (CNLS) fitting was used to analyse the impedance data.The fitting procedure is described elsewhere [73].This EEC consists of an electrolyte resistance(Rs),a time constant (characterized by a constant phase element (CPE),Qg,and a pore resistanceRg) related to the coating/electrolyte interface and another time constant (represented by a parallel combination of another CPE,Qdl,and a resistance,Rc) related to the metal/electrolyte interface.The incorporation of CPEs in the proposed EEC is justified by noting electrode porosity,distributed surface reactivity and roughness associated with this electrode geometry [74,75].Excellent agreement between the simulated and experimental impedance data(Fig.6b)confirms the validity of the proposed EEC.The error plots (a typical error plot is shown in the supporting information,Figure S5) reveal that maximum errors in the simulated data are ∼2% for the modulus of impedance(|Z|) and less than 3° for the phase angle.

Fig.6.(a) Proposed electrical equivalent circuit (EEC) fitted to the experimentally obtained impedance data of the RGO-CaCO3 coated specimen.(b) Bode plots of the experimental and simulated data for the RGO-CaCO3 coated specimen after 120 h of immersion in m-SBF.Temporal evolution of (c) Qg and Rg,and (d) Qdl and Rc,for the RGO-CaCO3 coated specimens after different immersion durations in m-SBF.
From the EEC it can be observed thatQgdecreases during the initial 25 h (Fig.6c),suggesting a decrease in the number densities of conductive pathways through the coating [61,70],which can be attributed to Hap formation that blocks the pores through the coating.This is further supported by the increasing pore resistance (Rg) during this initial immersion period(Fig.6c).Since increasingRgdecreases the exposure of the metal substrate to the electrolyte,this hinders a charge transfer process at the metal/electrolyte interface.This is also evident in the trend ofRc(Fig.6d),which attains its maximum at 25 h of immersion.The lowQgand highRgandRcexplain the highest corrosion resistance achieved at 25 h of immersion in the case of the RGO-CaCO3coating.However,after 25 h,Qgincreases,suggesting the development of conductive pathways through the coating [61,70],which indicates that after 25 h the Hap dissolution rate predominates the Hap formation rate.The predominance of the Hap dissolution process explains the decrease inRgafter 25 h,which facilitates easy electrolyte access to the metal substrate and thus enhances a charge transfer process at the metal/electrolyte interface.The enhanced charge transfer rate at the metal/electrolyte interface is confirmed by the decreasing trend inRc(Fig.6d).In fact,the increasingly exposed area of the metal/electrolyte interface due to the development of conductive pathways through the coating and consequent decrease in pore resistance explain the increase inQdl(Fig.6d) with increasing immersion time [28,45,73].The gradual increase inQgandQdl,plus decrease inRgandRc,also explain the slight decrease in the corrosion resistance of the RGO-CaCO3coated specimen with increasing immersion duration inm-SBF.
3.4. Mechanical integrity due to coating
With the presence of RGO in the RGO-CaCO3coating it is possible that apart from offering long-term corrosion protection,this coating may enhance the mechanical integrity/stresscorrosion cracking (SCC) resistance of the magnesium alloy in load-bearing conditions.The improved corrosion resistance is expected to suppress the electrochemical dissolution which is likely to improve resistance to SCC.Moreover,because graphene-based materials possess high toughness,the presence of RGO in the RGO-CaCO3coating is also likely to improve resistance of the coating to fracture,and thereby,improving resistance to SCC.The mechanical integrity of the RGO-CaCO3coating was evaluated by first immersing the coated tensile specimens inm-SBF for 120 h,followed by straining them in air at a strain rate of 3.1×10-7s-1.The details of this experiment are described in the supporting information section S8.The mechanical integrity of the coated specimen was compared with that of the uncoated one.The gauge lengths of the RGO-CaCO3coated and uncoated tensile specimens before and after immersion inm-SBF are shown in Fig.7a-d.At a macroscopic level,significant localised corrosion was evident in case of the uncoated specimens (as identified within an ellipse in Fig.7b),whereas the RGO-CaCO3coated specimens showed uniform degradation over the entire gauge length (Fig.7d).At the microscopic level,the morphology of the corroded RGO-CaCO3coated specimens was similar to that depicted in Fig.4c.The visible colour change in case of the RGO-CaCO3coated specimens can be attributed to the formation of different phases,such as hydroxyapatite,after 120 h of immersion in SBF.
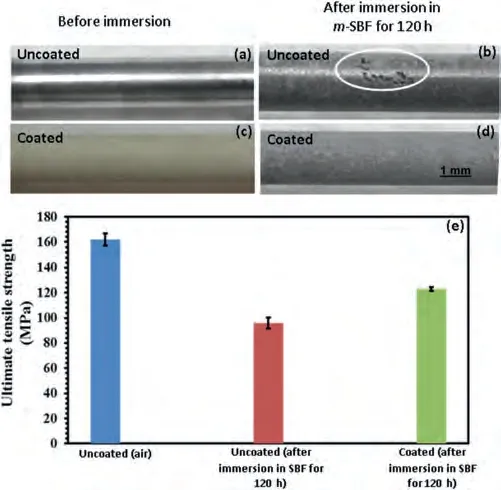
Fig.7.Photos of the gauge length of (a) and (b) uncoated,and (c) and (d)coated tensile specimens before and after immersion in m-SBF solution for 120 h,and (e) Ultimate tensile strengths for the uncoated specimen pulled in air,pre-immersed uncoated specimen pulled in air,and pre-immersed coated specimen pulled in air.
The ultimate tensile strength (UTS) data of the RGOCaCO3coated and uncoated specimens pulled in air at a strain rate of 3.1×10-7s-1(after immersion inm-SBF) are shown in Fig.7e.For comparison,the UTS of the uncoated specimen without exposure tom-SBF was also determined.The averaged UTS (126 MPa) of the coated specimen after immersion for 120 h inm-SBF was 18% higher than that of the uncoated specimen (96 MPa) after immersion for the same time.
Biodegradable implants are required to have good biocompatibility and desirable degradation performance.The RGOCaCO3coating not only provides excellent biocompatibility by generating Hap on the magnesium alloy surface,but also increases mechanical integrity.RGO-CaCO3coating,thus,provides a viable solution to circumvent the high corrosion rate of magnesium-based implants.
4.Conclusions
A thin RGO-CaCO3coating was deployed on an AZ91 magnesium alloy to improve its corrosion resistance and mechanical integrity inm-SBF.The corrosion resistance of the RGO-CaCO3coated specimen was an order of magnitude higher than that of the uncoated alloy and the only CaCO3coated specimen.Increasing immersion time inm-SBF until 25 h improved the barrier properties of the RGO-CaCO3due to in-situ formation of Hap on the coated surface,which also blocked pores present in the RGO-CaCO3coating.After 25 h of immersion,the dissolution process of Hap became more prominent compared to the formation process.As a result,the corrosion resistance of the RGO-CaCO3coating gradually decreased.However,even at 120 h of immersion,the corrosion resistance provided by the coating was about an order of magnitude higher than that of the uncoated alloy,and no significant damage was observed on the RGO-CaCO3coated specimen.Furthermore,the average UTS of the RGO-CaCO3coated specimen after immersion for 120 h in m-SBF was 18% higher than that of the uncoated AZ91 alloy after the same immersion time.RGO-CaCO3thus represents an effective biocompatible composite barrier coating for biodegradable magnesium implants.
Declaration of competing interest
I am writing to advise that authors of the manuscript as described below declare no conflict of interest.
CRediT authorship contribution statement
Lokesh Choudhary:Conceptualization,Data curation,Formal analysis,Investigation,Methodology.Parama Chakraborty Banerjee:Conceptualization,Formal analysis,Investigation,Methodology,Visualization,Writing– original draft,Writing– review &editing.R.K.Singh Raman:Conceptualization,Formal analysis,Funding acquisition,Project administration,Resources,Supervision,Writing– original draft,Writing– review &editing.Derrek E.Lobo:Investigation,Methodology.Christopher D.Easton:Investigation,Methodology.Mainak Majumder:Investigation,Resources.Frank Witte:Writing– review &editing.Jörg F.Löffler:Investigation,Writing– review &editing.
Acknowledgements
The authors gratefully acknowledge the facility and services of the Monash Centre for Electron Microscopy(MCEM)and the Melbourne Centre for Nanofabrication (MCN) in the Victorian Node of the Australian National Fabrication Facility(ANFF).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.jma.2023.12.012.
杂志排行

Journal of Magnesium and Alloys的其它文章
- A comprehensive review on the processing-property relationships of laser strengthened magnesium
- Recent advances in electrochemical performance of Mg-based electrochemical energy storage materials in supercapacitors: Enhancement and mechanism
- Peri-implant gas accumulation in response to magnesium-based musculoskeletal biomaterials: Reframing current evidence for preclinical research and clinical evaluation
- Influence of laser parameters on the microstructures and surface properties in laser surface modification of biomedical magnesium alloys
- Experimental and simulation research on hollow AZ31 magnesium alloy three-channel joint by hot extrusion forming with sand mandrel
- Mg/MgO interfaces as efficient hydrogen evolution cathodes causing accelerated corrosion of additive manufactured Mg alloys: A DFT analysis