基于综合生物信息分析鉴定心房颤动相关炎症基因及其与免疫细胞浸润的关联
2024-04-11赵兴安葛芸娜王玺雅陶四明
杨 曼 ,赵兴安 ,葛芸娜 ,秦 娟 ,王玺雅 ,陶四明
(1)大理大学临床医学院/大理市第一人民医院心内科,云南 大理 671000;2)云南大学附属医院/云南省第二人民医院/云南省眼科医院心内科,云南 昆明 650021)
心房颤动(atrial fibrillation,AF)增加心力衰竭和缺血性卒中的风险,并且对疾病相关的发病率和死亡率有显著贡献。AF 的发病机制非常复杂,一系列神经、体液、炎症反应、氧化应激等因素激活多种细胞因子和信号通路,最终导致心肌纤维化和心脏功能受损。然而,确切的病因和病理生理机制迄今尚未完全阐明。随着分子免疫学的发展与新兴技术的应用,许多基因表达谱已被应用于研究免疫细胞的分布和遗传生物标志物[1],为AF 的靶向预防和个体化治疗提供参考。
研究表明,活化的免疫细胞如中性粒细胞、单核细胞/巨噬细胞、肥大细胞、T 细胞、树突状细胞可能是组织修复、重塑和纤维化的关键参与者[2-3]。近年来的研究从免疫浸润的角度,提出了基于炎症反应的房颤发病机制的新见解[4]。然而,免疫炎症反应在房颤中的潜在机制仍然知之甚少。巨噬细胞是心脏免疫细胞中最重要的成员[2],对心脏稳态起着关键的调节作用。巨噬细胞与炎症密切相关[4],减少促炎巨噬细胞的数量或功能的治疗策略在一些临床研究中逐渐显现[5]。进一步探索炎症相关基因和免疫细胞,特别是巨噬细胞在AF 中的作用和机制将有助于临床工作者更好地理解该疾病。
在本研究中,一方面,笔者从综合数据集中筛选出AF 和窦性心律(sinus rhythm,SR)患者左心耳组织样本的差异表达基因(differentially expressed genes,DEGs);另一方面,笔者使用CIBERSORT 算法和加权基因共表达网络分析(weighted gene correlation network analysis,WGCNA)的组合方法来识别浸润免疫细胞的相关基因(immunerelatedgenes,IRGs)。随后,对2 种方法共享的基因(differentially expressed immune-related genes,DEIRGs)进行生物功能富集分析,并通过蛋白-蛋白相互作用(protein-protein interactions,PPI)网络分析、Cytoscape 软件筛选出候选基因。接着,笔者使用3 种机器学习算法构建预测模型,识别关键基因;并通过ROC 曲线验证关键基因的诊断效能,最后,使用Spearman 分析了关键基因与免疫细胞亚型之间的相关性。本研究旨在探讨免疫细胞在AF 中的浸润特征,寻找潜在的诊断和治疗靶点,为后续的临床研究提供指导。
1 资料与方法
1.1 微阵列数据的采集
下载的AF 和SR 基因表达谱数据及相应临床信息来自基因表达综合数据库(GEO,https://www.ncbi.nlm.nih.gov/geo/)。5 个包含人左心耳组织RNA含量的微阵列数据集(GSE14975、GSE31821、GSE79768、GSE41177 和GSE115574)被纳入进一步评估。以上5 个数据集均基于GPL570 平台,总共包含了来自 31 名SR 和44 名 AF 瓣膜病患者的左心耳组织样本。本研究中的所有数据均来自公共数据库,因此不需要获得该机构的伦理批准。生物信息学分析流程,见图1。
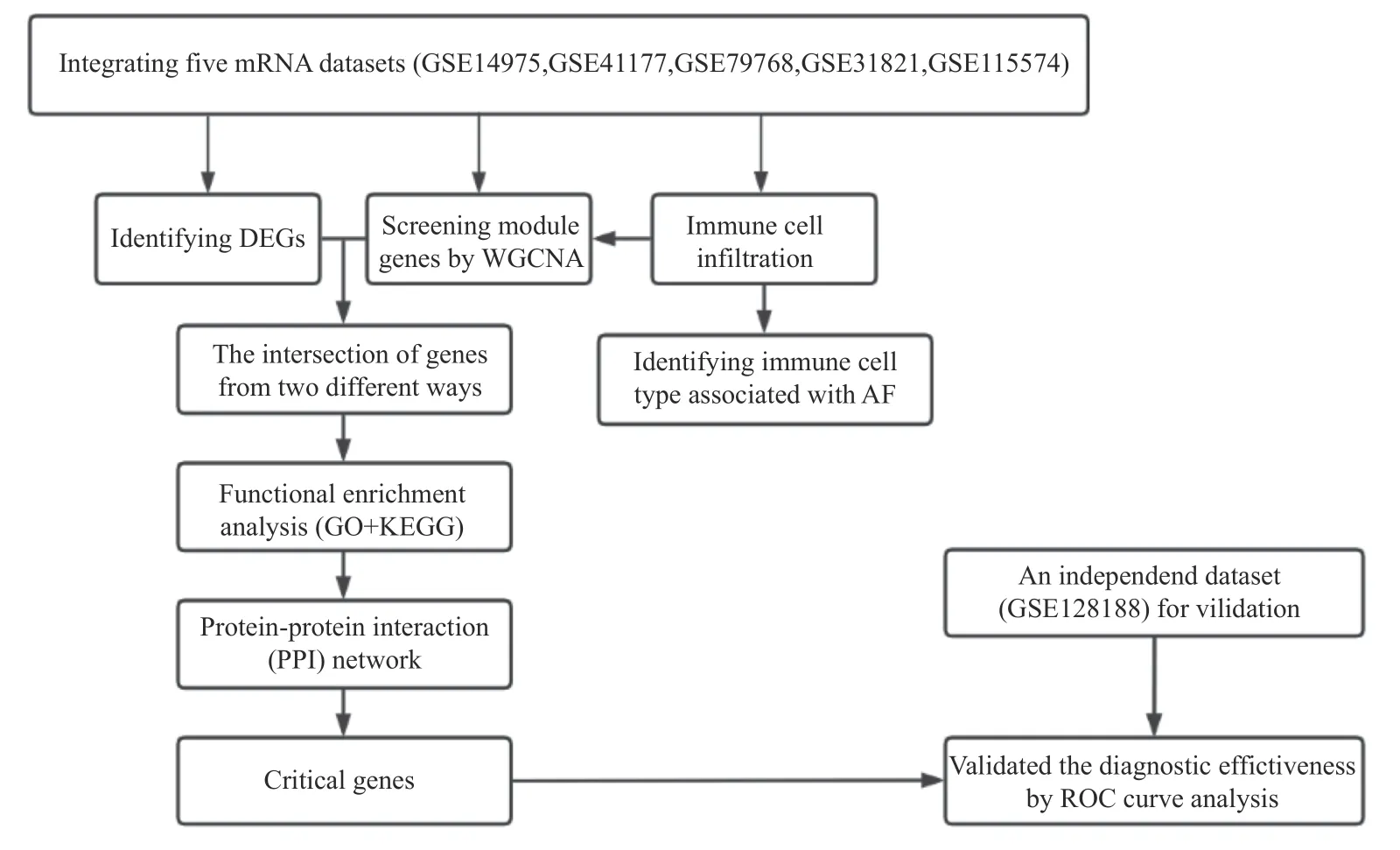
图1 生物信息学分析的流程图Fig.1 Flow diagram of the bioinformatics analysis
1.2 数据处理与DEGs 筛选
利用R(4.3.1)软件合并下载的5 个微阵列数据集,矩阵文件通过“sva”包标准化和去批次效应处理,并用主成分分析(principal component analysis,PCA)图可视化。探针ID 通过perl 语言(5.28.1)转换为基因符号,创建基因表达矩阵。AF 和SR 患者之间的DEGs 筛选采用R 软件的“limma”软件包进行。选择P<0.05 和| log2(fold change,FC)|>1 作为阈值。最后,根据上述数据使用“pheatmap”R 包绘制热图和火山图。
1.3 免疫细胞浸润分析
在R 中使用CIBERSORT 算法[6]对整合的基因表达矩阵文件进行免疫浸润分析,采用P<0.05对样本进行过滤,得到22 种免疫细胞亚型的浸润比例和相关性。“ggplot2”R 包和“ggpubr”R 包用于绘制细胞比例堆积图,AF 和SR 2 组间22 种免疫细胞亚型的浸润分数用R 中的“vioplot”包生成小提琴图。对22 种浸润性免疫细胞进行Spearman 相关分析,并采用“Corrplot”包生成相关热图。
1.4 WGCNA 识别免疫相关模块
加权基因共表达网络[7]使用R 软件构建,将表达模式相似的基因进行聚类,探讨基因表达与免疫细胞浸润之间的关系。综合数据集使用绝对中位差对基因进行降序排列,选择前10 000 个基因,选择软阈值来验证无尺度网络。基于拓扑重叠矩阵(topological overlap matrix,TOM)的差异度对平均连锁层次进行聚类,然后将表达模式相似的基因聚类为模块,采用平均连锁层次聚类方法,用不同的颜色进行标记。每个模块的最小基因数为30 个,模块合并的阈值为0.25。根据免疫浸润的特征,选择P值最小、相关性最高的模块作为关键模块。关键模块中的基因(IRGs)被筛选出用做下一步分析。
1.5 基因本体论(gene ontology,GO)和京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)富集分析
取DEGs 和IRGs 交集的基因(DEIRGs)在维恩图[Draw Venn Diagram(ugent.be)]中呈现。利用注释、可视化和富集分析数据库(DAVID,2021-Update,https://david.ncifcrf.gov/home.jsp)对DEIRGs进行GO 和KEGG 富集分析。研究模块基因的生物学功能包括生物过程(biological processes,BP)、细胞成分(cellular components,CC)、分子功能(molecular functions,MF)和信号通路,P值<0.05和计数≥3 被认为显著富集。
1.6 蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络的构建
在STRING(https://cn.string-db.org)网站中将DEIRGs 导入检索工具,生成PPI 网络,评估蛋白质之间相互作用关系。评分>0.4 识别基因间相互作用的阈值。节点代表蛋白质,边代表PPI 网络中的蛋白质-蛋白质关联。然后使用Cytoscape软件(v3.8.1)对从STRING 数据库中下载的结果进行可视化。Cytoscape 软件的MCODE 插件来识别枢纽基因,基于MCC(maximum clique centrality)算法从得分排名筛选出候选基因。
1.7 机器学习算法识别AF 的生物标志物
利用最小绝对值收敛和选择算子算法(least absolute shrinkage and selection operator,LASSO)、随机森林(random forest,RF)、支持向量机递归特征消除(support vector machine recursive feature eilmination,SVM-RFE)3 种机器学习算法在R 中使用“mlr3verse”、“randomForest”和“e1071”包构建AF 的预测模型。对每个预测模型的特征重要性进行分析和排序,筛选出关键基因。具体而言,首先,基于“mlr3”R 包,候选基因的表达数据根据7∶3 的比例随机分配到训练集和测试集。接着,使用交叉验证筛选出最佳的变量组合。最后,使用“pROC”R 包绘制ROC 曲线。获得ROC 曲线下的面积(area under curve,AUC)值和95%的置信区间(confidence interval,CI)值,以评估关键基因的敏感度和特异度。
1.8 关键基因的诊断及预测价值分析
为了验证关键基因的诊断有效性和预测价值,一方面,关键基因在综合数据集中的差异表达用“ggpubr”R 包绘制箱线图,并用“glm”函数进行Logistic 回归分析,构建AF 诊断模型,评价关键基因的预测价值。另一方面,从GEO 数据库中下载了包含10 个左心耳组织样本的独立数据集GSE128188(AF=5,SR=5,GPL18573 平台),采用“pROC”R 包建立ROC 曲线,计算AUC 值和95%CI值验证关键基因的诊断效能。
1.9 生物标志物与浸润免疫细胞的相关性分析
在R 中导入关键基因在综合数据集中的表达矩阵和免疫浸润分析结果文件,使用“corrplot”包中的“Spearman”方法对关键基因和浸润免疫细胞进行相关性分析,结果通过“ggpubr”包可视化。
2 结果
2.1 AF 中DEGs 的鉴定
将5 个数据集合并后的基因表达矩阵进行标准化,批次效应去除后的结果用二维PCA 聚类图呈现(图2A),PCA 图表明,2 组样本的聚类明显,说明样本来源是可靠的。在44 个AF 和31 个SR样本中,使用 “limma”包共鉴定出593 个DEGs,其中387 个表达上调,216 个表达下调,见图2B和图2C。
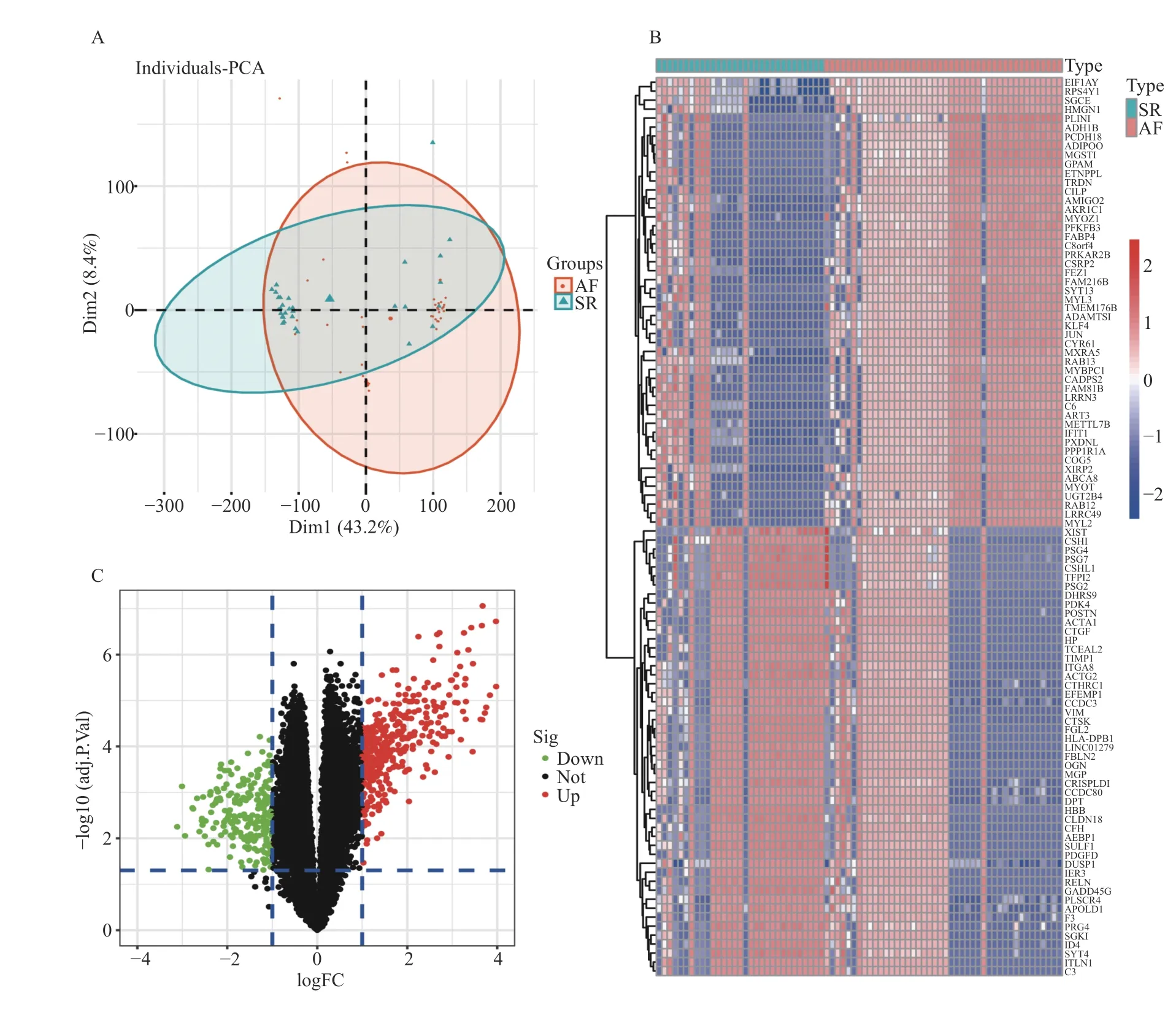
图2 AF 和SR 样本组间的DEGs 鉴定Fig.2 Identification of DEGs between AF and SR samples
2.2 免疫浸润分析
使用CIBERSORT 算法分析了综合数据集中免疫细胞的浸润水平。图3A 说明了来自44 个AF 样本和31 个SR 样本的免疫细胞比例。图3B表示7 种免疫细胞亚型的浸润丰度在2 组样本间有差异(P<0.05),与SR 相比,来自AF 患者的左心房组织样本含有更高的活化的CD4 记忆T 细胞、M1 巨噬细胞、M2 巨噬细胞、活化的肥大细胞,而原始的B 细胞、静息的树突状细胞、静息的肥大细胞含量更低。22 种免疫细胞相关性分析表明,M2 巨噬细胞与单核细胞的正相关关系最强(r=0.51),而M2 巨噬细胞与M0 巨噬细胞呈明显负相关(r=-0.82),见图3C。
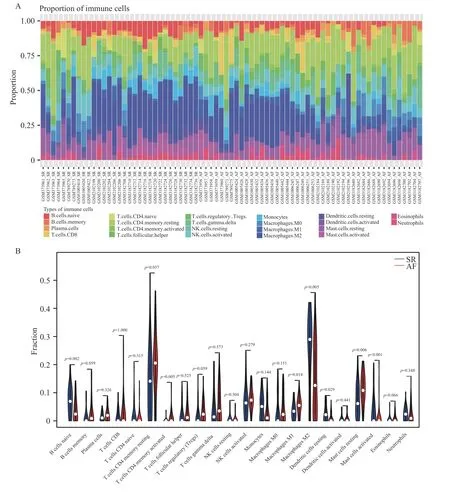
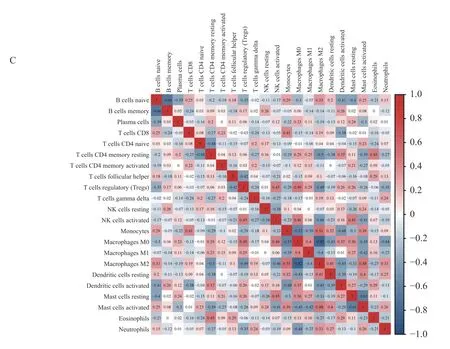
图3 免疫细胞浸润的分布和相关性Fig.3 Distribution and correlation of immune cell infiltration
2.3 WGCNA 鉴定浸润免疫细胞的关键模块
为了进一步阐明AF 相关免疫细胞的潜在机制和功能,在R 中使用“WGCNA”包,构建无尺度分布网络(图4A),选择软阈值β=10 来验证,结果显示R2=0.81,slope=-0.5,符合无尺度网络的标准。采用分层聚类的方法将网络划分为多个模块,模块合并后共获得4 个模块(图4B)。免疫浸润的特征和模块的相关性热图表明,黑色模块与M2 巨噬细胞、静息的树突状细胞、活化的肥大细胞呈正相关,而与静息的肥大细胞呈负相关,见图4C。选择黑色模块作为关键模块,其中包含3 441 个IRGs,见图4D。
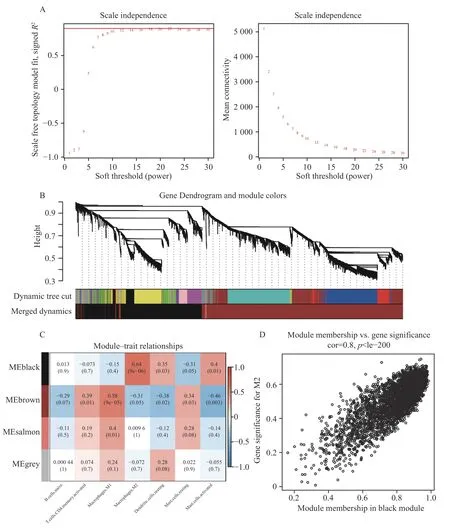
图4 WGCNA 分析Fig.4 WGCNA analysis
2.4 DEIRGs 的功能富集分析
对DEGs 和IRGs 共享 的190 个DEIRGs 进 行GO 和 KEGG 分析以了解这些基因的生物学功能和富集信号通路。BP、CC、MF 显示了10 个最显著的结果(图5A),最与免疫反应(如体液免疫反应、补体激活)、免疫细胞(髓系白细胞分化的负调控作用)和免疫活动(MHCII 类蛋白复合物)有关。KEGG 通路富集分析表明,这些DEIRGs 主要在扩张性心肌病、肥厚性心肌病、病毒性心肌炎、风湿、系统性红斑狼疮(systemic lupus erythematosus,SLE)等疾病中富集,并参与吞噬、同种异体移植物的排斥、抗原处理和呈递、细胞粘附等免疫功能,同时在TGF-β 信号通路富集,见图5B。
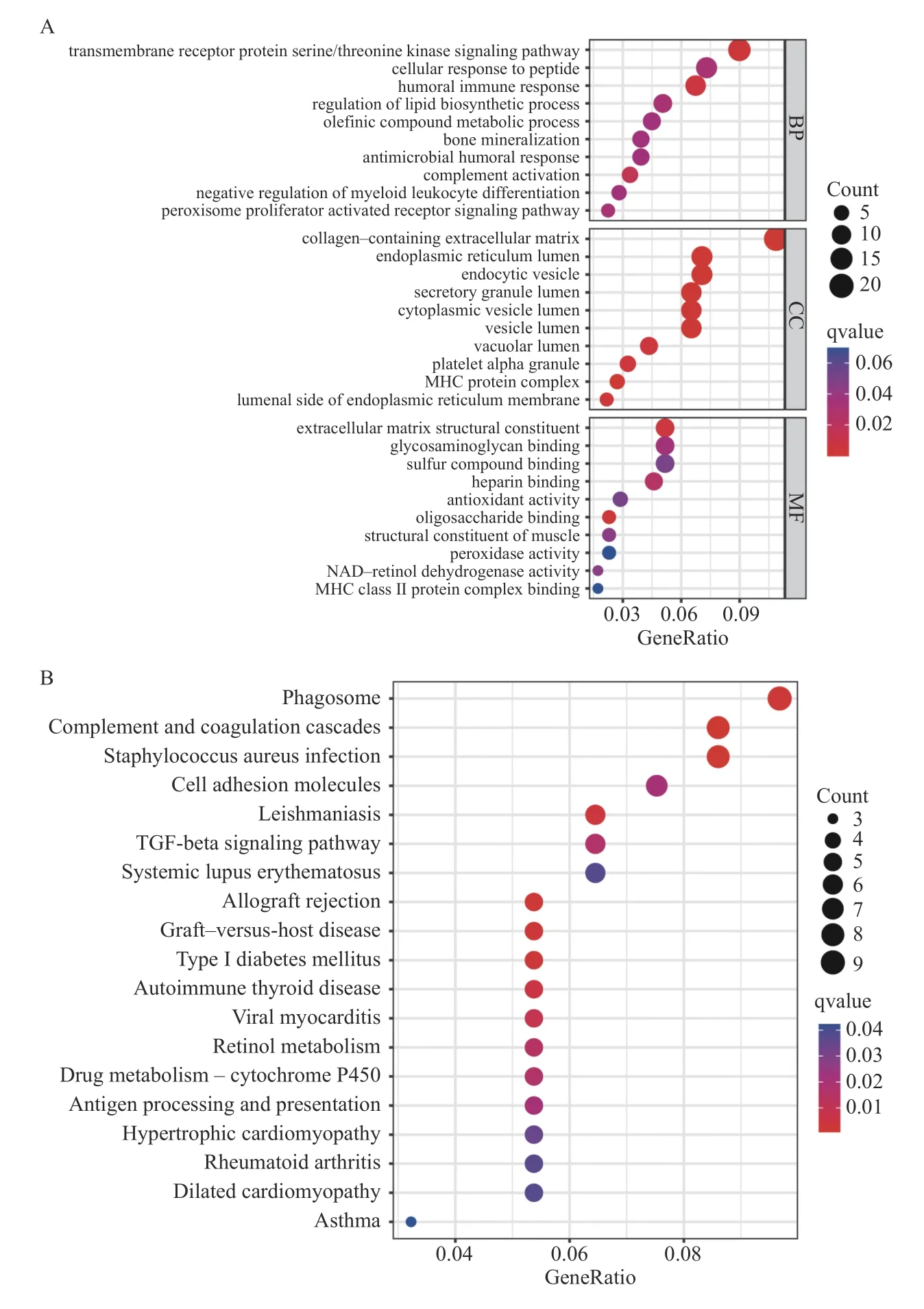
图5 DEIRGs 的GO 和KEGG 功能富集分析Fig.5 GO and KEGG functional enrichment analysis of the DEIRGs
2.5 PPI 网络筛选候选基因
在3 441 个IRGs 中,共有190 个基因与DEGs重叠,见图6A。笔者使用 STRING 数据库构建了一个由190 个DEIRGs 相交的PPI 网络,去除孤立节点后,使用CytoScape 构建了包含131 个节点和284 条边的PPI 网络,见图6B。节点之间的高互联性在 PPI 网络表明了蛋白质之间的功能内聚性。此外,MCC 算法确定了前10 个候选基因。这些候选基因的排名,见图6C。
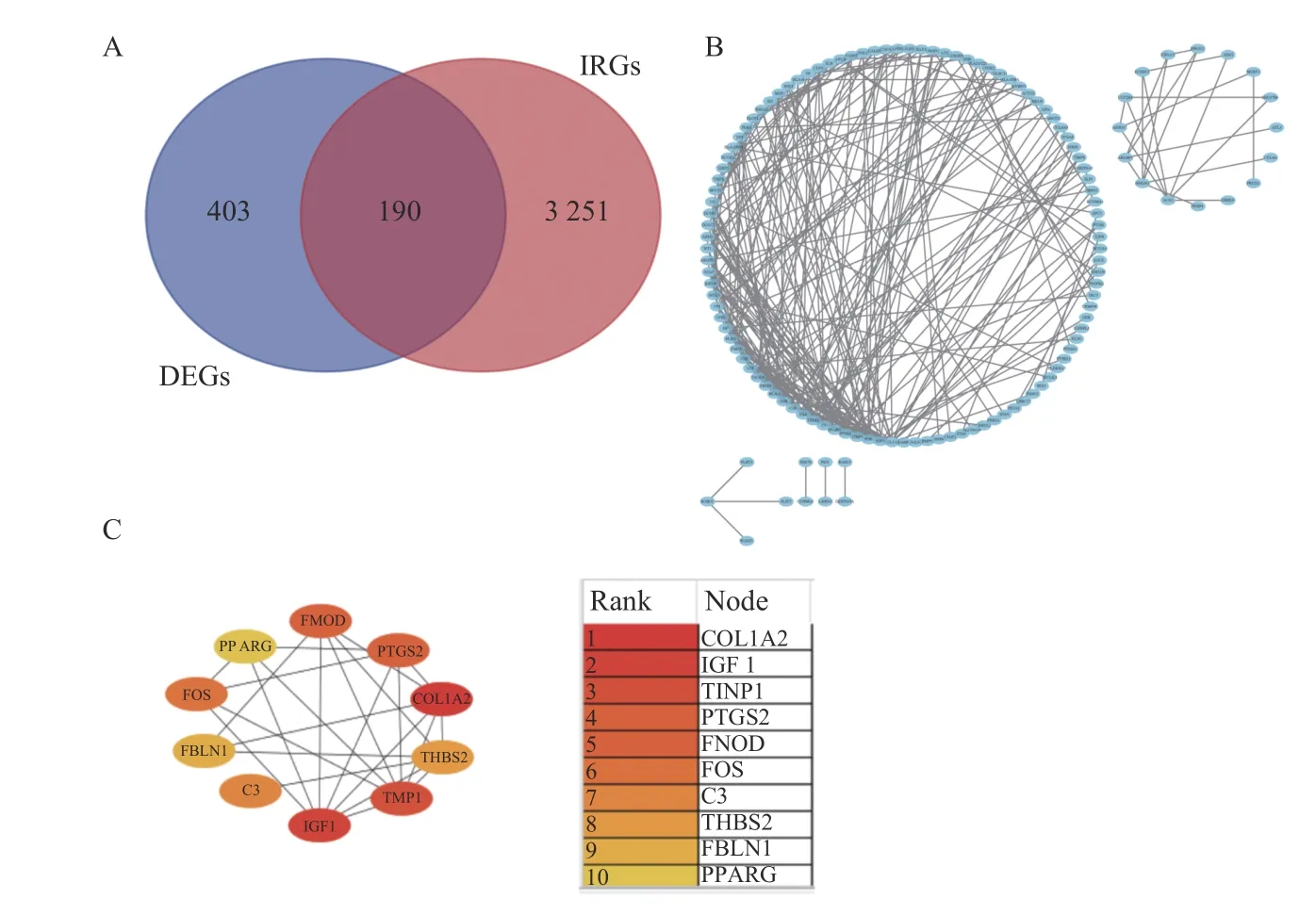
图6 PPI 网络筛选候选基因Fig.6 PPI network screening for candidate genes
2.6 机器学习算法预测AF 生物标志物
为了获得较高的准确性,应用3 种机器学习算法从10 个候选基因(COL1A2、IGF1、TIMP1、PTGS2、FMOD、FOS、C3、THBS2、FBLN1 和PPARG)中筛选出核心基因。使用 LASSO 回归算法降低了候选基因数量,确定了3 个关键基因作为预测模型(IGF1、PTGS2 和PPARG),见图7A;在RF 算法中笔者选择MeanDecreaseGini 值大于2 的基因作为预测模型(C3、IGF1、PPARG、TIMP1、THBS2、PTGS2 和COL1A2),见图7B;SVM-RFE 算法确定了5 个基因作为预测模型(PPARG、TIMP1、PTGS2、IGF1 和C3),见图7C。ROC 曲线验证了3 种算法的预测效果(AUC,95%CI),见图7D。最终,笔者选择了预测效果最好的LASSO 回归算法中的的3 个关键基因(IGF1、PTGS2、PPARG)作为AF 的潜在生物标志物,笔者发现这3 个基因同时也在3 种算法的预测模型中共享。

图7 机器学习算法识别 AF 生物标志物Fig.7 Machine learning algorithm identifies AF biomarkers
2.7 预测模型有效性评价与验证
为了评估3 个标志物的预测价值,使用箱线图展示了3 个关键基因在综合数据集中的表达情况,结果显示在AF 组中IGF1、PTGS2 和PPARG的表达量高于SR 组,见图8A。接着,基于3 个关键基因的表达情况笔者构建了列线图作为诊断模型,如图8B 所示,总的评分越高,AF 的风险就越高。此外,在另一个独立的数据集(GSE128188)中,3 个关键基因的诊断有效性使用ROC 曲线得到进一步验证,PPARG 的AUC 为0.880(95%CI=0.626~1),IGF1 的AUC 为0.760(95%CI=0.372~1),以及PTGS2 的AUC 为0.72(95%CI=0.365~1),见图8C~8E。
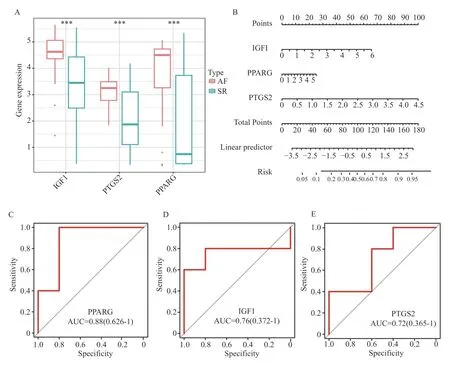
图8 生物标志物的诊断及预测效能Fig.8 Diagnostic and predictive efficacy of the biomarkers
2.8 生物标志物与浸润免疫细胞的相关性分析
为了进一步说明3 个关键基因与浸润免疫细胞的相关性,分析结果用棒棒糖图可视化,图9A~9C 分别代表IGF1、PTGS2 和PPARG 和22 种免疫细胞亚型的相关性和P值。

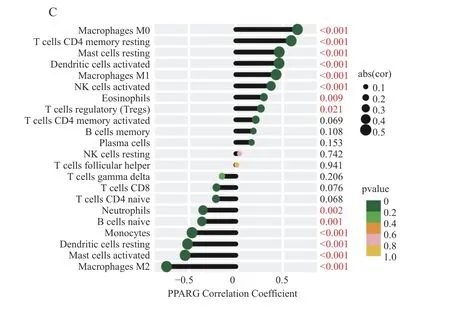
图9 3 个关键基因与22 种免疫细胞亚型的相关性分析Fig.9 Correlation analysis of three key genes with 22 immune cell subtypes
3 讨论
免疫炎症反应在许多心脏病理生理过程中起着重要的作用,包括AF 导致的心肌损伤和纤维化修复过程[8]。然而,由于免疫炎症反应在房颤发生和发展过程中的复杂性和多样性,它们在AF 发病机制中的作用仍需进一步研究。据报道,一些炎症相关基因与AF 密切相关,如趋化因子受体CXCR2,可能在心房重构和AF 诱导过程中发挥重要的作用[9];基因SPP1 通过与局部免疫细胞和基质细胞的串扰促进心房颤动[10]。基于免疫细胞浸润特征,分析炎症生物标志物及其信号分子可能有助于预测患者AF 的风险[11]。
笔者的分析结果表明,AF 的进展与免疫细胞的浸润密切相关。在本研究中,采用 CIBERSORT 算法探索AF 中免疫细胞的浸润特征,发现炎症相关免疫细胞在AF 组中比例增多,并且M2巨噬细胞与单核细胞和M0 巨噬细胞相关性最强。巨噬细胞是心脏中最丰富的白细胞,是细胞因子的主要来源,参与许多重要的免疫、炎症反应、内环境稳态和代谢过程[11]。研究发现,AF 患者左心耳组织中单核细胞和巨噬细胞是最具有免疫活性的[12]。M2 巨噬细胞极化后可表现出一些与M1 的重叠特征[13],具有促炎的作用。这些结果说明巨噬细胞是心脏的重要免疫细胞,在炎症刺激后巨噬细胞的极化状态在维持心脏的稳态和心房重塑过程中发挥着重要作用。笔者的研究进一步证明并强调了这些标记的重要性。
DEIRGs 是AF 免疫炎症过程中的特征基因。基于WGCNA 的结果,笔者提取了与M2 相关性最强的黑色模块基因,并筛选出与DEGs 共享的190 个DEIRGs,这些DEIRGs 的GO 分析结果主要涉及免疫细胞、免疫活动和免疫反应过程。研究表明,体液免疫反应、补体激活、髓系白细胞分化的负调控、受体配体活性和细胞粘附等生物过程和分子功能与房颤的进展密切相关[14-15]。MHCII 类蛋白复合物作为巨噬细胞移动抑制因子(macrophage migration inhibitory factor,MIF)的主要受体,协同MIF 促进心房炎症和纤维化[16]。KEGG 结果主要在扩张性心肌病、肥厚性心肌病、病毒性心肌炎、风湿、SLE 和TGF-β 信号通路富集。众所周知,心肌病是AF 的基础,可能与遗传、免疫相关。另外,研究表明,SLE 可能是房颤的独立危险因素[17],在AF 中,TGF-β 是促进心房纤维化发生和维持的重要信号通路[18]。笔者的结果说明了这些DEIRGs 参与了AF 的免疫和炎症过程。
IGF1、PPARG 和PTGS2 被识别为AF 的炎性生物标志物。通过构建PPI 网络和基因显著性评分,笔者筛选出了10 个候选基因。据报道,其中一些基因已被证明与房颤发病机制有关[19-21],这说明了本结果的可靠性。通过机器学习算法和ROC 曲线分析,3 个 关键基因(IGF1、PTGS2 和PPARG)最终被确定为AF 相关生物标志物。研究表明,IGF1 具有促生长活性,调节心脏的收缩、新陈代谢、肥大、自噬、衰老和细胞凋亡,与心血管疾病风险升高有关[22]。在炎症刺激下,IGF1作为IGF1R(IGF-1 receptor)的配体,激活酪氨酸激酶的活性,从而激活PI3K-AKT、MAPK 等下游信号通路[23]。先前的研究表明,PI3K-AKT[24]、MAPK[25]信号通路介导心房重构,在房颤发生、发展过程中发挥作用。PTGS2 是1 种前列腺素生物合成中的关键酶,可以激活巨噬细胞的吞噬作用[26],在炎症反应中具有特殊作用[27]。一项心肌细胞试验证明CV-3 可能通过调节PTGS2 治疗房颤[28]。PPARG 是1 种蛋白编码基因,与PPARG相关的疾病包括胰岛素抵抗、糖尿病和高血压[29]。研究发现,PPARG 通过抑制NF-kappa-B 介导的促炎反应,作为肠道稳态的关键调节器[30]。房颤患者的心肌重塑与环形时钟基因BMAL1 的表达减少有明显的相关性[31],而PPARG 可通过调节血管中BMAL1 的转录来调节心血管昼夜节律[32]。这些研究结果表明,3 个关键基因参与了房颤的病理生理过程,和炎症密切相关。
IGF1、PPARG 和PTGS2 与浸润免疫细胞关系密切。相关性分析结果表明,3 个关键基因的表达与静息CD4 记忆T 细胞、M0 巨噬细胞呈显著正相关,而与M2 巨噬细胞呈显著负相关,基于这些发现,笔者推测IGF1、PPARG 和PTGS2可能通过调节免疫细胞浸润在AF 的发生发展过程中发挥作用。然而,这些结果需要额外的体内或体外实验来证明关键基因和免疫细胞浸润之间的复杂相互作用。
综上所述,本研究通过综合生物信息学分析鉴定了3 个与AF 炎症相关的生物标志物(IGF1、PTGS2 和PPARG),笔者还对这些基因与特异性免疫细胞的关系进行了分析与评价。这些结果可能为AF 的免疫系统浸润模式提供新的见解。
