一种快速测定酱油和醋中对羟基苯甲酸酯的方法
2024-04-10杨姗姗赵海峰王晓雯
◎ 杨姗姗,赵海峰,英 瑜,王晓雯,金 伟
(青岛市食品药品检验研究院,国家药品监督管理局海洋中药质量研究与评价重点实验室,山东 青岛 266071)
对羟基苯甲酸酯又称羟苯甲酯或尼泊金酯,因其存在酚羟基结构,可破坏细胞膜,使细胞内的蛋白质变性,抑制微生物的酶系统活性,造成微生物的新陈代谢异常,从而阻止其正常生长和繁殖,具有抑菌作用[1];又因其晶体无色或白色、无味,不会影响产品的色泽和风味,在人体内代谢迅速、不会产生累积,所以被国际公认为安全、高效、广谱的防腐剂,在食品工业、化妆品、医药等领域得到了广泛使用。随着研究的深入,1978 年,ISHIDATE 等[2]研究表明对羟基苯甲酸酯可以引发染色体畸变;1998 年,ROUTLEDGE 等[3]发现对羟基苯甲酸酯存在雌激素活性;2002 年,日本东京都卫生研究所报道,即使微量摄入对羟基苯甲酸丁酯,也会造成实验大鼠精子数量和浓度的显著下降,对雄性小鼠生殖系统造成不良影响[4];2008 年,DARBRE 等[5]的研究发现,对羟基苯甲酸酯具有雌激素和抗雄激素活性,不仅会影响男性生殖功能,还有可能增加乳腺癌的发病率;2012 年,SUGANDHA 等[6]证实对羟基苯甲酸酯确实会增加女性患乳腺癌的风险。对羟基苯甲酸酯的激素活性被越来越多的人所重视,在食品工业中的应用也受到了一定的限制。
目前,我国要求酱油、醋中对羟基苯甲酸酯类及其钠盐(以对羟基苯甲酸计)最大使用量均为0.25 g·kg-1[7],食品中对羟基苯甲酸酯的检测方法主要有气相色谱法[8-11]、气相色谱-质谱法[12-15]、液相色谱法[16-19]、液相色谱-质谱法[20-22]、毛细管电泳法等[23]。GB 5009.31—2016 是目前食品中检测对羟基苯甲酸酯类的主要方法[24],但是操作烦琐,消耗时间长、试剂用量大,不利于大批量样品的检测工作。本文研究建立了测定酱油、醋中对羟基苯甲酸酯类样品前处理方法,该方法前处理工作操作简单,检测结果准确可靠,在实验室检测中既能提高工作效率,又可减少试剂用量,适宜大批量样品的检测工作。同时,本文的研究内容也对其他品类样品的前处理方法具有指导意义。
1 材料与方法
1.1 材料与试剂
4种对羟基苯甲酸酯类混合标准溶液(1 000 mg·L-1):购自国家标物中心;乙酸乙酯:HPLC 级,德国Merk公司;NaCl、NaHCO3:均为分析纯,国药集团化学试剂有限公司;实验用水均为Millipore 现制水。
1.2 仪器与设备
7890B 气相色谱仪:美国Agilent;Multi Reax 涡旋振荡器:德国Heidolph;3-18KS 高速冷冻离心机:美国Sigma;电子天平:美国Ohaus。
1.3 方法
1.3.1 溶液配制
饱和NaCl 的饱和NaHCO3溶液:在饱和NaHCO3溶液中加入NaCl 至过饱和。
饱和NaHCO3的饱和NaCl 溶液:在饱和NaCl 溶液中加入NaHCO3至过饱和。
1.3.2 样品前处理
称取5 g(精确至0.001 g)均匀样品于50 mL 离心管中,加入10 mL 乙酸乙酯,振摇3 min 后,5 000 r·min-1离心5 min,取上清液于新的50 mL 离心管中,加入10 mL 饱和NaCl 的饱和NaHCO3溶液,振摇3 min 后,5 000 r·min-1离心5 min,取5 mL 上清液,氮吹至近干,加1 mL 乙醇复溶,过0.22 µm 有机滤膜,待测。
1.3.3 色谱条件
色谱柱:HP-5(美国Agilent,30 m×0.32 mm,0.25 μm);载气:氮气;恒流模式:2 mL·min-1。
进样口温度:250 ℃;进样模式:不分流;进样体积:1 µL。
程序升温:100 ℃保持1 min;20 ℃·min-1升温至170 ℃;12 ℃·min-1升温至220 ℃,保持1 min;10 ℃·min-1升温至250 ℃,保持8 min。
FID 检 测 器 温 度:280 ℃; 空 气 流 速:450 mL·min-1;氢气流速:40 mL·min-1。
2 结果与分析
2.1 盐酸酸化步骤的去除
在样品前处理过程中加入1 ∶1 盐酸1 mL 对样品进行酸化,可以将对羟基苯甲酸的钠盐酸化为对羟基苯甲酸,但是对羟基苯甲酸的沸点为336.2 ℃,不易被汽化,需经甲酯化后方能使用气相色谱法进行测定[10]。若不进行甲酯化,由于对羟基苯甲酸的酸度比碳酸强,使用饱和碳酸氢钠洗涤样液时,样液中的对羟基苯甲酸会与碳酸氢钠反应再次生成对羟基苯甲酸钠盐,因此原方法中加入1 ∶1 盐酸1 mL 对样品进行酸化,对检测结果无影响。本文使用含有对羟基苯甲酸甲酯和对羟基苯甲酸乙酯的酱油样品就是否需要酸化样品进行了6 个平行样的对比,结果如表1 所示。在样品前处理过程中加入1 ∶1 盐酸对样品酸化和不进行酸化,测定结果无显著差异。另外,考虑到节俭、环保的要求,本文方法不再进行样品酸化。

表1 样品酸化比对实验结果表
2.2 萃取试剂的选择
在实验用水和18%的NaCl 溶液(模拟酱油的含盐量)中加入终浓度均为10 mg·L-1的4 种对羟基苯甲酸酯类,选择常用试剂甲醇、乙醇、乙醚、乙酸乙酯和乙腈,分别进行萃取。实验过程中发现,甲醇和乙醇均能分别与实验用水和NaCl 溶液互溶,无分层现象发生,不适宜作为对羟基苯甲酸酯的萃取试剂;乙腈可以与实验用水互溶,但与NaCl 溶液混匀后仍分层现象明显,因此在提取过程中可以通过盐析作用得到乙腈层,乙腈可以用作对羟基苯甲酸酯的萃取试剂;乙醚和乙酸乙酯与实验用水和NaCl 溶液混匀后均分层现象明显,可以用作对羟基苯甲酸酯的萃取试剂。
按1.3.2 的操作经离心、氮吹、复溶、过膜后测定,结果如图1 所示,用乙醚和乙酸乙酯提取实验用水中的对羟基苯甲酸酯类,上机检测,采用乙酸乙酯萃取时,4 种对羟基苯甲酸酯的加标回收率基本在105%左右,明显优于乙醚;用乙醚、乙酸乙酯和乙腈提取18% NaCl 溶液中的对羟基苯甲酸酯类,使用乙酸乙酯萃取时,4 种对羟基苯甲酸酯的加标回收率基本在100%~105%,整体上优于乙醚和乙腈。因此,最终选择乙酸乙酯作为萃取试剂。

图1 不同提取溶剂的对羟基苯甲酸酯类回收率图
乙酸乙酯提取18%NaCl 溶液中对羟基苯甲酸酯类的回收率相对提取实验用水的回收率偏低,可能是由于乙酸乙酯在实验用水和18% NaCl 溶液中的溶解度不同,溶于实验用水中的乙酸乙酯相对NaCl 溶液更多一些,造成乙酸乙酯层的体积减小,在溶质基本不变的情况下,溶剂体积减小,必然导致浓度的增高。
2.3 净化溶剂的选择
在5 g 酱油样品中加入10 mg 4 种对羟基苯甲酸酯的标准物质,使用乙酸乙酯提取,分别采用20 mL 10 g·L-1的NaHCO3溶液、20 mL 饱和NaCl 溶液、10 mL饱和NaCl 溶液+10 mL 10 g·L-1NaHCO3溶液、10 mL 10 g·L-11的NaHCO3溶液、10 mL 饱和NaCl 溶液、5 mL饱和NaCl 溶液+5 mL 10 g·L-1NaHCO3溶液对提取液进行净化。
实验过程中发现,提取液经含有NaHCO3的溶液净化后,有机层中的深色杂质会转移至水相中;若净化液中无NaHCO3,深颜色的杂质仍留存在有机层中,说明NaHCO3对提取液中的深色杂质有良好的去除作用,净化溶液中应含有NaHCO3。净化后的有机层,按1.3.2 的操作氮吹、复溶、过膜后测定,结果如图2 所示。使用20 mL 10 g·L-1的NaHCO3溶液净化提取液后,4 种对羟基苯甲酸酯的加标回收率大多高于110%,对羟基苯甲酸乙酯的加标回收率高于120%,过高的回收率说明20 mL 10 g·L-1的NaHCO3溶液不适宜用作本实验的净化。同时,对比10 mL 10 g·L-1的NaHCO3溶液净化,净化溶液浓度相同,加入的体积不同导致加标回收率明显差异,再次验证了乙酸乙酯在水溶液中的溶解量增加,会导致加标回收率的上升。同理,对比20 mL 饱和NaCl 溶液和10 mL饱和NaCl 溶液净化后的加标回收率,对比10 mL 饱和NaCl 溶液+10 mL 10 g·L-1NaHCO3溶液和5 mL 饱和NaCl溶液+5 mL 10 g·L-1NaHCO3溶液的加标回收率,净化溶液体积越大,其加标回收率相对偏高,因此净化过程中净化溶液的体积应控制在合理范围内。
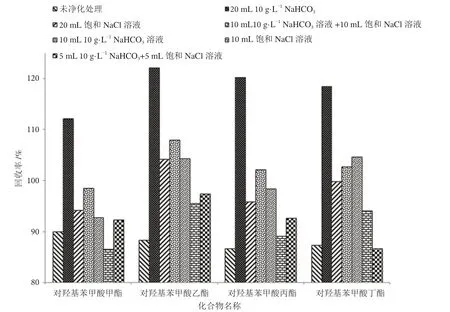
图2 不同净化溶液的对羟基苯甲酸酯类回收率图
前期的实验已经证明NaHCO3对去除提取液中杂质具有重要的作用,而且净化溶液的使用量会影响实验的最终结果。为了进一步验证净化溶液体积相同,不同的溶质组合对加标回收率的影响,实验室比对饱和NaCl 的饱和NaHCO3溶液,饱和NaHCO3的饱和NaCl 溶液,饱和NaCl 的10 g·L-1NaHCO3溶液的净化效果。分别取10 mL 净化溶液净化10 mL 提取液,提取液中的深色杂质都能转移至水相中。通过图3 的加标回收率比较,采用饱和NaCl 的饱和NaHCO3溶液净化后,4 种对羟基苯甲酸酯的综合回收率相对更优。
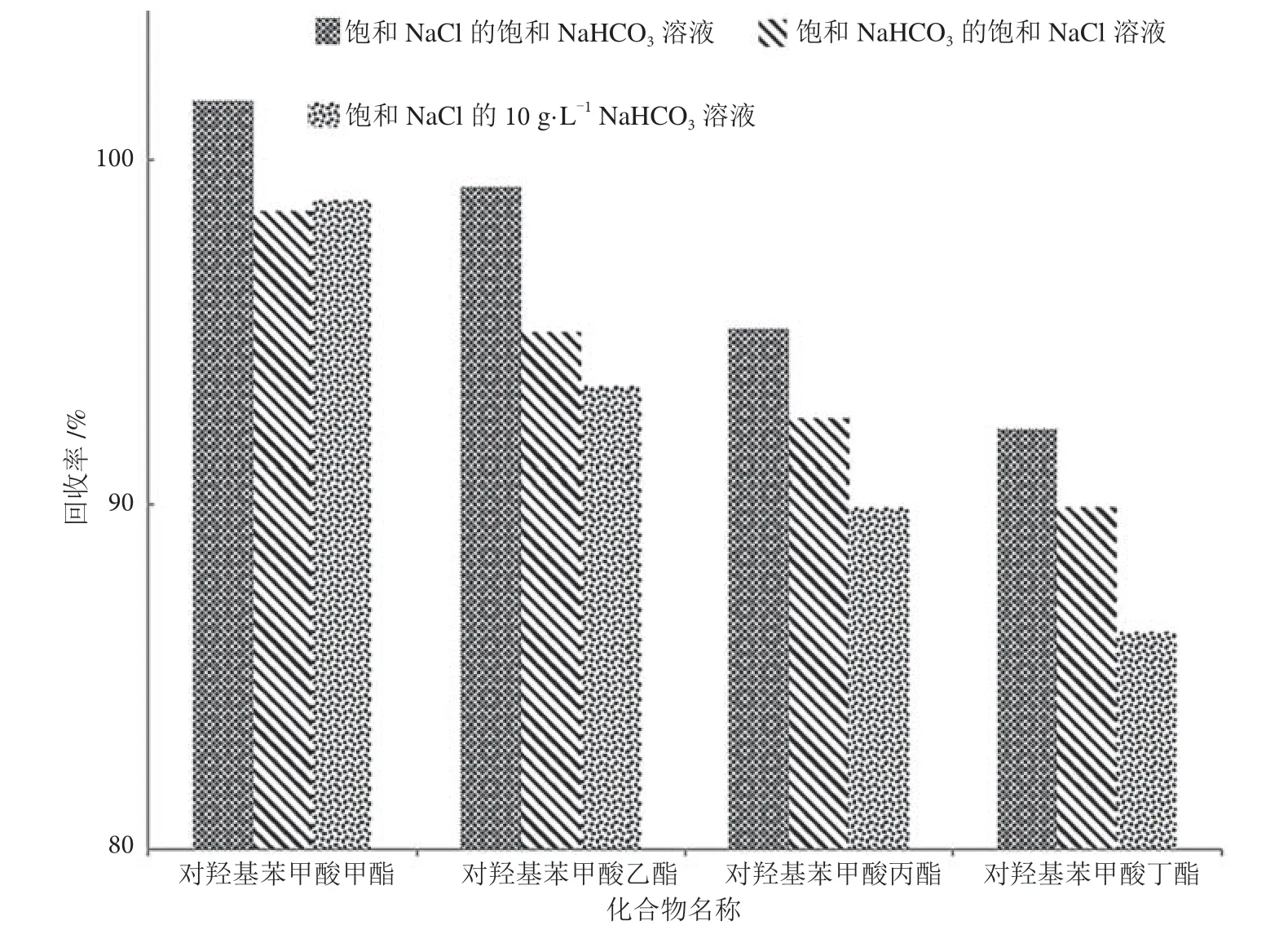
图3 不同净化溶液的对羟基苯甲酸酯类回收率图
2.4 标准曲线、检出限、准确度及精密度
用乙醇配制4 种目标物混标系列浓度4.0 mg·L-1、10.0 mg·L-1、20.0 mg·L-1、50.0 mg·L-1、100 mg·L-1、200 mg·L-1和300 mg·L-1进行测试,分别以4 种对羟基苯甲酸酯类的峰面积A为纵坐标,以质量浓度ρ(mg·L-1)为横坐标绘制标准曲线,详见表2。

表2 4 种目标物的线性关系、相关系数表
在未检出对羟基苯甲酸酯类的酱油样品和醋样品中,分别添加6 平行的0.6 mg·kg-1(GB 5009.31—2016的检出限)、2.0 mg·kg-1、4.0 mg·kg-1和10.0 mg·kg-1的对羟基苯甲酸酯,按照1.3.2 进行样品前处理,检测结果详见表3。4 种目标物在不同添加水平的回收率基本在70%~120%,相对标准偏差在0.90%~7.06%,表明该方法的准确度和精密度均较好。按照国标方法检出限水平添加,也能准确定量,能满足日常检测酱油、醋中对羟基苯甲酸酯类的要求。
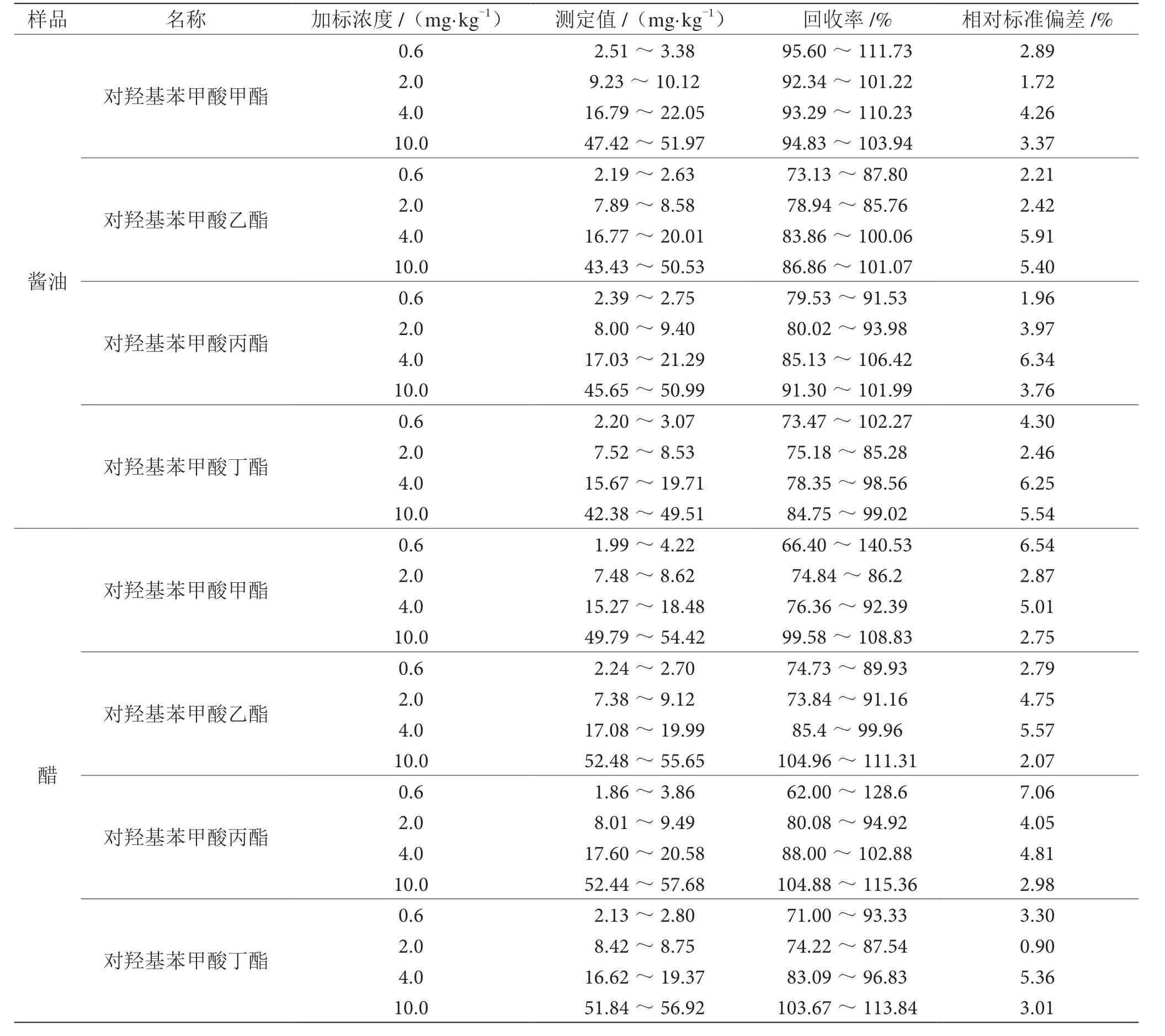
表3 方法回收率和精密度实验表(n=6)
2.5 讨论
本文样品前处理方法可以准确、快速高效、经济环保地测定酱油、醋中的对羟基苯甲酸酯类,单个样品的前处理时间约30 min,批量样品的前处理时间与单个样品的区别主要在于试剂添加的耗时,同时有机试剂用量少,检出限也满足GB 5009.31—2016 的要求。然而存在的问题是,GB 2760—2014 中的限量要求是针对“对羟基苯甲酸酯类及其钠盐”,而其指定检测方法GB 5009.31—2016 为仅能测定“对羟基苯甲酸酯类”,本文样品前处理方法也未考虑“对羟基苯甲酸钠盐”的测定,将在之后的研究中加以完善。
3 结论
对羟基苯甲酸酯是食品中常见的防腐剂,为保证食品质量安全,建立一种快速高效、经济环保的检测方法十分必要。本文研究了测定对羟基苯甲酸酯类的样品前处理的操作机理、优选了萃取试剂和净化方法,并使用实际样品验证了方法的检出限、准确度、精密度,建立了快速高效、经济环保测定酱油和醋中对羟基苯甲酸酯类的样品前处理方法,适宜大批量样品的快速测定。同时,本文的研究内容也对测定其他品类样品中对羟基苯甲酸酯类的前处理方法具有指导意义。
