仲醇聚氧乙烯醚硫酸钠的表面性能研究
2024-03-29张政委李晓东李玉霞
李 萍 张政委 康 锦 李晓东 李玉霞
晋中学院化学化工系高值精细化学品研究中心,山西晋中,030619
脂肪醇聚氧乙烯醚硫酸钠是由脂肪醇醚经三氧化硫硫酸化、氢氧化钠中和后所得的一类阴离子表面活性剂[1]。以伯醇醚为原料制得聚氧乙烯醚硫酸盐(AES)的制备技术在国内已经非常成熟,由于其产品性能优越[2],目前已被广泛应用于清洗剂、匀染剂、渗透剂等领域和个人护理产品配方中[3-4],属于第二大类阴离子表面活性剂。2021年其在中国国内产、销量达到77.54万吨和76.31万吨,占当年阴离子表面活性剂总产、销量的50.84%和51.69%[5]。由于AES的上游原料进口依赖度较高,其价格受马来西亚、印度尼西亚油脂产出国贸易和政策影响较为明显,因此开发本土原料资源制备脂肪醇醚硫酸钠具有重要的现实意义。
仲醇及其衍生物具有良好的表面活性,在液体洗涤剂、纺织印染、农药乳化等领域有广泛的应用[6-10]。仲醇聚氧乙烯醚硫酸盐(SAES)由于其特殊的分子结构而具有优越的耐碱性及渗透能力等性能,因而其物化及应用性能的研究也备受关注。SAES的主要原料仲醇在合成技术上有较大的难度,目前市场上的仲醇及其衍生物仲醇醚主要来自日本触媒化学和美国陶氏化学,江苏赛科是目前国内唯一实现仲醇工业化生产的企业。因此,目前国内有关仲醇醚硫酸钠的研究鲜有报道。
本文主要对SAES的光学微流变性、倾点、平衡和动态表面张力、铺展性等物化、表面和应用性能进行了研究,并与AES的性能进行了对比,以丰富SAES的理论研究,并为其在应用领域的实际应用提供相关理论数据。
1 实验部分
1.1 试剂与仪器
化学试剂:仲醇聚氧乙烯醚(SAE,烷基链长C12-14,EO数为3,江苏赛科化学有限公司),仲醇聚氧乙烯醚硫酸钠(SAES,烷基链长C12-14,EO数为3,实验室自制,制备方法如文献[11]所示),伯醇聚氧乙烯醚硫酸钠(AES,烷基链长C12-14,EO数为3,中轻化工股份有限公司)。为了对SAES和AES进行更好的对比,分别对上述硫酸盐产品加入无水乙醇除无机盐,滤液收集后用石油醚多次萃取,除去未硫酸化物、水相经旋蒸除水后得到黄色黏稠状物。
根据GB/T 5173-2018 《表面活性剂 洗涤剂阴离子活性物含量的测定 直接两相滴定法》对其阴离子活性物含量进行测定,根据GB/T 6366-2012《表面活性剂 无机硫酸盐含量的测定滴定法》对其无机硫酸盐含量进行测定,根据GB/T 13530-2023《乙氧基化烷基硫酸钠试验方法》对其未硫化物含量进行测定。其中,SAES中阴离子活性物含量为92.6%,无机硫酸盐含量为0.06%,未硫化物含量为1.9%,其余为水分;AES中阴离子活性物含量为93.1%,无机硫酸盐含量为0.05%,未硫化物含量为1.7%,其余为水分。
实验仪器:Rheolaser Master光学微流变仪(法国Formulaction公司),K-12表面张力仪(德国KÜRSS公司),BP100型最大泡压法表面张力测定仪(德国KÜRSS公司),DSA25型接触角测量仪(德国KÜRSS公司),Turbiscan Tower稳定性分析测试仪(法国Formulaction公司)。
1.2 物化性能测定
1.2.1 光学微流变
使用Rheolaser Master光学微流变仪对质量浓度为30%的SAES和AES进行黏弹性测定。将20 mL液体样品放入微流变仪专用样品池中,25 ℃下扫描2 h进行测定。弹性因子(elasticity index,EI)和宏观黏度因子(macroscopic viscosity index,MVI))由Rheosoft Master1.4.0.1软件记录。
1.2.2 平衡表面张力
用二次蒸馏水配制一定浓度的SAES和AES溶液,静置过夜后待用。在(25±0.1)℃时,采用连续吊环法测定其表面张力。二次蒸馏水的表面张力在(72.0±0.3)mN/m范围之内,每次测试前都要先测量二次蒸馏水的表面张力来检验样品池内是否残留表面活性物质。cmc由表面张力对浓度曲线的转折点确定[12]。
1.2.3 动态表面张力
用二次蒸馏水配制不同浓度的SAES和AES溶液,静置过夜后待用。采用德国KÜRSS公司的最大泡压法表面张力测定仪测定各样品在不同浓度下的动态表面张力曲线,测定温度为(25±0.1)℃,测量的有效表面时间为10~200 s。
1.2.4 铺展性
用二次蒸馏水配制不同浓度的SAES和AES,静置过夜后待用。在(25±0.1)℃时,采用DSA25型接触角测量仪,通过对不同浓度样品水溶液在石蜡膜上的动态接触角的测定对其铺展性进行分析。
1.2.5 润湿性
参考GB/T 11983-2008《表面活性剂 润湿力的测定 浸没法》,采用帆布沉降法对样品的润湿性进行测定。将1 g/L样品溶液倒入1 000 mL烧杯中,在25 ℃时,把直径为30 mm的圆帆布片用浸没夹夹住,浸入试样液,帆布片进入溶液时立即开始计时,帆布片开始下沉时记为终点。每个样品重复测试3次,取平均值[13]。
1.2.6 起泡性和稳泡性
使用稳定性分析测试仪对SAES和AES进行起泡性和稳泡性测定。将1 g/L的20 mL样品放入测量池内,25 ℃时每25 s扫描一次,共扫描3 h进行测定。
1.2.7 乳化性
采用量筒法[14]对样品的乳化性能进行测定。将1 g/L表面活性剂水溶液40 mL和等体积的液体石蜡置于100 mL具塞量筒中,上下剧烈振荡5次,静置1 min,重复5次,记录分出10 mL水所需的时间。每个样品重复测试5次,取平均值。
1.2.8 耐盐耐碱性
取1 mL的1 g/L样品溶液于10 mL比色管中,加入一定浓度(固定浓度最大为300 g/L)不同体积的NaCl/NaOH溶液(梯度可选1 mL,2 mL,3 mL,4 mL,5 mL,6 mL,7 mL,8 mL,9 mL),最后补加去离子水,保持总体积量为10 mL。将比色管上下倒置10次(每次控制在2 s左右),25 ℃下静置2 h,观察比色管内溶液的浑浊程度。采用UV-1601型紫外可见分光光度计测试不同盐/碱浓度样品溶液在600 nm处的透光率,透过率>80%的盐/碱浓度(g/L)即为该表面活性剂浓度下的最大耐盐/碱度。
2 结果与讨论
2.1 光学微流变分析
微流变可以通过跟踪粒子的布朗运动获得均方位移(mean square displacement, MSD)值测量样品的黏弹性特性[15]。从图1中可以看出,随着样品老化时间的增加,SAES和AES的MSD曲线都是首先由长位移向短位移转变,即弹性增加,粒子间碰撞增加;去相关时间由短向长的MSD曲线变化,表明二者的黏度增加。SAES和AES在样品老化过程中,其结构都发生了变化,黏弹性都有所增加。从图1中还可以看出,SAES和AES的黏弹性差别较大,在老化过程中SAES黏弹性变化较小,而AES的黏弹性变化较大。

图1 SAES与AES的均方位移曲线(a)SAES;(b)AES。
图2(a)为SAES和AES的弹性因子(EI)随时间变化曲线。从图中可以看出,随着时间的增加,AES的弹性因子逐渐增大,说明在样品放置过程中其结构明显增强;而SAES的弹性因子基本不发生变化,说明在样品放置过程中其结构基本不发生变化。这可能是因为AES的亲水基SO4-基团位于碳氢链的端点,而SAES的SO4-基团位于疏水链的中间位置,相比AES,SAES结构中存在支链,粒子间相互作用较差。AES的最终弹性因子为1.77×10-2nm-2,明显高于SAES的最终弹性因子9.10×10-4nm-2。

图2 SAES与AES的弹性因子(a)和宏观黏度因子(b)随时间变化曲线
图2(b)为SAES和AES的宏观黏度因子(MVI)随时间变化曲线。粒子移动到一定距离的时间越长,表示液滴运动速度越低,MVI值越高,相应地样品的黏度越高,体系的结构越强。从图中可以看出,随着时间的增加,SAES和AES的宏观黏度因子都有所增加,但增加的幅度不大。AES的最终宏观黏度因子为4.21×10-2nm-2·s,远远高于SAES的最终宏观黏度因子6.91×10-5nm-2·s。在实际应用过程中,黏度更低的SAES使用更为方便。
2.2 表面张力
2.2.1 平衡表面张力
图3为SAES和AES水溶液的表面张力随浓度变化曲线。从图中可以看出,随着SAES和AES水溶液浓度的增加,表面张力都逐渐降低,这意味着在界面上开始出现紧密排列,到达cmc以后,表面张力不再变化,为一常数,说明在体相内已经形成了聚集体。根据公式1和公式2可以得出表面活性剂在气液界面的饱和吸附量Гmax和最小分子截面积Amin。根据Dahanayake等[16]提出的吸附效率pC20的物理意义,即得溶剂表面张力降低20 mN/m时所需表面活性剂浓度的负对数,根据公式3可以计算出表面活性剂的pc20值。pc20是度量表面活性剂表面活性的一个重要参数,其值越高说明表面活性剂的吸附效率越高。

图3 25 ℃时SAES和AES水溶液的表面张力随其浓度的对数变化曲线
式中n是一个常数,对于1∶1型离子型表面活性剂,其在水溶液中以表面活性离子和反离子的形式存在,根据电中性原则,正离子与负离子的吸附量相等,式中n=2。R为气体常数8.314 J/(mol·K),T为绝对温度K,NA为阿伏伽德罗常数6.02×1023,C20为当水的表面张力降低20 mN/m时所需表面活性剂的浓度。由表面张力曲线得到AES和SAES的表面性质相关数据如表1所示。从表1可以看出,AES溶液的cmc和γcmc分别为0.45 mmol/L和30.77 mN/m,该值与文献报道的AES的cmc(0.33 mmol/L)和γcmc(33~34 mN/m)数值接近[17-18]。SAES的cmc和γcmc分别为0.42 mmol/L和27.71 mN/m。SAES和AES的cmc值基本一致,这可能是由于二者具有相同的碳原子数,分子之间的疏水相互作用基本相同所致。相比AES,SAES由于支化结构的存在,增加了表面活性剂分子在水表面的覆盖率,从而使γcmc有所降低。这与一些文献报道的支链型表面活性剂降低表面张力的能力优于直链型的结果相一致[19-21]。支链化的结构也使其Amin增大,Гmax减小。

表1 25 ℃时SAES和AES的表面活性参数
2.2.2 动态表面张力
对某一浓度表面活性剂溶液,达到吸附平衡前某一时刻的表面张力称为动态表面张力(dynamic surface tension, DST),在某些体系中动态表面张力比平衡表面张力更重要。如在涂膜过程中,新表面不断形成,若表面张力降低速度缓慢,就会导致涂布不均匀等。因此研究表面张力随时间的变化具有十分重要的意义。本实验通过最大泡压法分别测定了25 ℃下两种表面活性剂在水溶液中的吸附行为。结果如图4所示。

图4 25 ℃时不同浓度的SAES和AES水溶液的动态表面张力(a)SAES;(b)AES。
从图4可以看出所测不同浓度不同表面活性剂的表面张力在初始阶段均快速下降,一定时间后趋于平稳状态。这是因为动态表面张力测试过程中新形成的表面远偏离于平衡态,吸附层离子的浓度很低,体相与吸附层的浓度差大,因此动态表面张力变化很大。随着吸附的进行,次表面浓度和表面浓度逐渐增大,扩散和吸附的速度均逐渐降低,动态表面张力随着吸附时间的增加而降低的趋势也逐渐变得平缓[22]。当表面活性剂浓度增大时,表面张力的下降幅度和速度也随之增大,而且相应地缩短了到达平衡状态的时间。这种随体系浓度的增加达到平衡所需的时间越短的趋势与已经报道的一些烷基表面活性剂的结果是相一致的[23-25]。在相同浓度下,SAES表面张力下降的速率大于AES,这是由于SO4-在碳氢链中间的化合物比在端点者扩散速度快[26]。
当溶液浓度较小时,可以采用Ward-Tordai方程来描述溶液体相与次表面之间的传质过程[27]:
从该公式中可以看出后一项含有卷积积分,使此方程不可解。
当t→0时,属于短时吸附过程,由公式4可转化为公式5[28]:
式中,γt指时间t时的表面张力(mN/m);γ0为去离子水的表面张力(mN/m);SAES和AES为1∶1型阴离子表面活性剂,则n=2,Ds为短时吸附过程的扩散系数(m2/s);R为气体常数8.314 J/(mol·K),T为绝对温度K。
当t→∞时,此过程属于长时吸附过程,可由公式4转化为公式6:
结合Gibbs方程,可演变成公式7:
式中,γeq为t→∞时的表面张力(mN/m);Г为表面过剩量;Гeq为平衡表面过剩量;Dl为长时吸附过程的扩散系数(m2/s)。
图5和图6分别为不同浓度的SAES溶液和AES溶液的γt随t1/2与t-1/2的变化曲线图。分别对其进行拟合,当t→0时,γt与t1/2呈直线关系,直线的截距为溶剂水的表面张力;当t→∞时,γt与t-1/2也呈直线关系,直线的截距为不同浓度下表面活性剂水溶液的平衡表面张力。通过求直线部分的斜率可以得出Ds、Dl值及相应的比值。结果列于表2中。由表2可以看出,随浓度的增加,Ds与Dl均减小,这是因为随着浓度的增大,分子间静电斥力作用增大,从而使得分子的自由运动受到限制。通过比较SAES和AES的Ds值可以发现,SAES的Ds值均大于AES,说明SAES在吸附初期的吸附扩散速率较快;SAES的Dl值均小于AES,说明SAES在吸附后期由于空间位阻等的效应使得吸附势垒更高。Ds与Dl的比值远小于1,这意味着Ds与Dl差别比较大,说明吸附后期为混合动力控制吸附[29-30]。

表2 25 ℃时不同浓度的SAES和AES水溶液的扩散系数及其比值

图5 25 ℃时不同浓度的SAES和AES水溶液的动态表面张力随t1/2的变化(a)SAES;(b)AES。

图6 25 ℃时不同浓度的SAES和AES水溶液的动态表面张力随t-1/2的变化(a)SAES;(b)AES。
2.3 铺展性
2.3.1 动态接触角
研究表面活性剂溶液在固体表面的铺展性可为其在涂覆、化妆品等领域的应用提供相应的理论支持[31]。SAES和AES水溶液各自在不同浓度下的接触角(θ)变化曲线如图7所示。由图7可知,随着SAES与AES浓度的增加,θ值逐渐减小,且随着时间的延长,相对较高浓度下的θ值越小。在溶液浓度远低于cmc时,SAES和AES的动态接触角随时间延长减小很少,而在溶液浓度接近cmc时,SAES和AES的动态接触角随时间延长而明显减小。表面活性剂水溶液在石蜡膜上的润湿过程可以理解为表面活性剂分子在液/固界面的吸附过程。根据表面张力梯度理论[32],在表面活性剂分子的扩散过程中,液滴的前沿和体相之间产生界面张力梯度差,促进表面活性剂溶液在固体界面扩散,浓度越高,扩散速度越快。随着时间的增加,体相中表面活性剂分子数量减少,导致界面张力梯度差减小,使扩散速度减慢,最终趋于平衡。从图中还可以看出在溶液浓度接近cmc时,SAES溶液接触角小于AES接触角,说明SO4-在碳氢链中间的SAES在石蜡表面的扩散速度快、铺展性能较好,该结果与动态表面张力测试结果相一致。

图7 25 ℃时不同浓度的SAES和AES水溶液在石蜡膜上的动态接触角(a)SAES;(b)AES。
2.3.2 铺展系数
铺展是固-液界面取代固-气界面同时增大气-液界面的过程,铺展系数(S)是描述溶液对固体润湿性能的一个重要参数,S值越大,铺展性能越好,润湿效果越好[33]。S的计算公式如下:
式中,γsg、γsl和γlg分别为固-气、液-固和气-液的界面张力,θ0为液滴在低能表面上的平衡接触角。由图3可得不同浓度下SAES与AES水溶液的γlg值;对图7所得的不同时间的接触角采用指数衰减函数进行曲线拟合,获得平衡接触角θ0,指数衰减函数如公式9所示[34]。
公式(9)中,θ为接触角,θ0为平衡接触角,A为润湿缓冲系数,T为润湿时间常数,t为时间。不同浓度下SAES与AES水溶液的γlg值及其在石蜡表面的θ0值见表3。通过公式8计算可得SAES与AES的铺展系数随摩尔浓度变化的关系图,如图8所示。从图中可以看出,对于SAES和AES,二者的S都随浓度的增加而逐渐增加,说明溶液的浓度越高,SAES和AES的铺展性越强。这是由于表面活性剂分子疏水基团以疏水相互作用吸附于固体表面,亲水基团朝向体相,使表面亲水化导致接触角减小,随着表面活性剂浓度的增大,表面活性剂分子在固-液界面上的吸附量增大,此时,表面张力的降低与接触角减小之间的协同效应使得铺展系数增大。相同浓度时,SAES的S值略大于AES,说明相对于AES,SAES在石蜡表面的铺展性能较好。

表3 25℃时不同浓度的SAES和AES水溶液的γlg值及其在石蜡表面的θ0
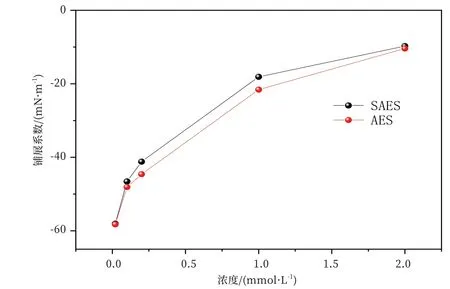
图8 SAES与AES的铺展系数随浓度变化的关系图
2.3.3 黏附张力
黏附张力(γlgcosθ)体现了固体与液体之间的黏附能力。研究发现表面活性剂在疏水表面润湿时的黏附张力与其水溶液的表面张力之间存在如下线性关系[35]:
式中,a和b为常数,θ为平衡接触角。SAES和AES的黏附张力与表面张力的关系如图9所示。从图9可以看出,黏附张力随着表面张力的增大而逐渐减小,在较低浓度时,随着吸附量的增加,表面张力降低,黏附张力增大,表面活性剂分子在气-液界面和固-液界面逐渐形成单分子吸附层。在较高浓度下,表面张力降低的趋势减小,黏附张力却急剧增加,说明表面活性剂分子气-液界面吸附逐渐要达到饱和,但在固-液界面的吸附仍在继续进行。在整个浓度范围内黏附张力与表面张力不存在线性关系,但γlgcosθ~γlg的关系图可分为两部分,这两部分均存在良好的线性关系。这是因为在较高浓度下,SAES和AES的表面活性剂分子穿过液滴落在石蜡表面上降低了石蜡的表面张力,同时引入的聚氧乙烯基团与固体表面发生极化作用,两方面原因的共同作用使得石蜡的表面性质发生了变化[36]。这与文献报道的部分表面活性剂在低能表面的铺展润湿行为相一致[33,37]。

图9 SAES与AES的黏附张力与表面张力的关系图
Lucassen-Reynders根据杨氏方程和Gibbs方程得到了应用于研究界面相对吸附的经验公式[38]:
式中,Гsg、Гsl和Гlg分别代表表面活性剂在固-气、固-液和气-液界面的吸附量。通常由于表面活性剂分子溶于水中,固-气界面不存在吸附,因此Гsg=0。通过小于cmc之前直线部分拟合的斜率可以得到Гsl和Гlg的比值。由图9可知,SAES和AES的a值分别为-0.68和-0.37,即这两种表面活性剂在石蜡表面的值均小于1,说明其在石蜡-水界面的吸附量小于其在气-液界面的吸附量,与SAES相比,AES的a绝对值更小,说明AES在固-液界面的吸附更小于气-液界面。
2.4 起泡性和稳泡性
在表面活性剂的各种应用领域中,对其泡沫性能要求并不相同。沐浴露和洗涤剂等清洁产品追求较高的起泡性能,但在大规模工业清洗和印刷行业中则需要低泡沫性能[39]。表面活性剂的泡沫性能通常从起泡能力和稳泡能力两个方面来评价[40]。图10中,其横坐标表示泡沫的高度,横坐标越大说明样品的起泡性越强。如图10所示,SAES的泡沫高度为10.36 mm,AES的泡沫高度为15.56 mm,说明AES的起泡性优于SAES,这是因为与SAES相比,AES具有较好的表面活性,起泡性强。从图10中还可以看出,对于SAES和AES,其透射光的ΔT(%)图谱右边有一个光强值上升的峰,说明样品池的顶部有澄清层,并且随着时间的增加已经有光能够透过了,即溶液中泡沫逐渐破裂,样品池顶部有空气导致光能够透过;其透射光的ΔT(%)图谱左边有一个光强值上升的峰,说明样品池此位置有光能够透过,即泡沫破裂后得到的水逐渐增加导致光能够透过;其背散射光光强值ΔBS(%)图谱有整体向下的趋势,说明随着时间的增加泡沫的粒径逐渐增加导致光强值逐渐降低。

图10 SAES与AES的背散射光光强值和透射光光强值图谱(a)SAES;(b)AES。
体系的泡沫稳定性可以用稳定性动力学指数来表征,稳定性动力学曲线的计算公式如公式12所示:
稳定性动力学指数(di)反映的是样品在整个放置时间浓度和颗粒粒径的变化幅度的综合值,稳定性动力学指数越大,系统越不稳定。图11为SAES与AES的稳定性动力学曲线,从图中可以看出,在开始的0.5 h内,SAES与AES的稳定性动力学指数几乎相同,但随着时间的增加,SAES的稳定性动力学指数明显高于AES,说明在开始的0.5 h内,二者的泡沫稳定性几乎相同,但是随着时间的延长,SAES体系的稳泡性明显弱于AES,这是由于带有一定支链的SAES分子在气-液界面上形成的表面膜黏度低,不够紧密结实,泡沫稳定性较差[19]。

图11 SAES与AES的稳定性动力学曲线
2.5 乳化性
乳化作用是在一定的外力作用下使不混溶的两种液体形成具有一定稳定性的液液分散体系的作用,该分散体系被称为乳状液,是一种热力学不稳定体系[41]。为了评估SAES和AES的乳化性,采用量筒法测定了二者对液体石蜡的乳化能力,记录从乳液中分离出10 mL水所需要的时间,时间越长,乳化性能越好。SAES与AES的乳化性对比如表4所示。从表中可以看出,SAES的乳化性低于AES,这可能是因为SAES分子中的支链结构,使得其分子总体积相对较大,在空间位阻以及电性斥力作用使疏水基难以插入油相中,即与液体石蜡之间的相互作用较弱,乳化性较差。

表4 SAES与AES的乳化性对比
2.6 润湿性
润湿性是表面活性剂的基本应用性能之一。帆布沉降法测定的是表面活性剂溶液对棉布的润湿效果,润湿过程的快慢取决于表面活性剂的性质。由表5可知,SAES、AES对帆布片的润湿时间分别为26 s和60 s,表明SAES的润湿性优于AES,这是由于SAES结构中存在支链,表面活性剂分子从水溶液中迁移到气-液界面的速率较快,容易扩散到纤维表面,降低帆布片表面张力的能力较强,从而具有较好的润湿性[19]。

表5 SAES与AES的润湿性对比
2.7 耐盐耐碱性
加入一定量的盐/碱可以提高离子型表面活性剂在洗涤及采油等应用中的界面活性[42],而盐/碱浓度达到一定数值时,会导致表面活性剂从水溶液中析出而降低其表面活性,因此耐盐/碱性较好的表面活性剂有助于扩大其应用范围[43]。从图12中可以看出,SAES耐NaCl盐度约为250 g/L,而AES在300 g/L NaCl盐溶液中透光率数据均在80%及其以上,后者耐NaCl盐性更好;SAES耐NaOH碱度约为30 g/L,AES耐NaOH碱度为约60 g/L,后者耐碱性更好,即SAES的耐盐耐碱性均低于AES。
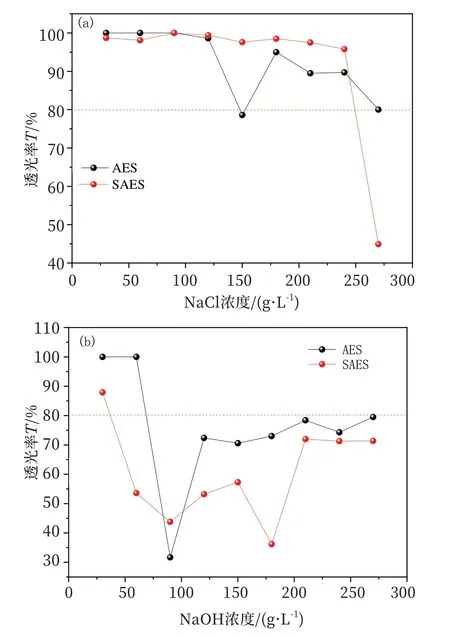
图12 SAES和AES溶液的透光率随NaCl和NaOH浓度的变化
3 结论
通过研究仲醇聚氧乙烯醚硫酸钠(SAES)和伯醇聚氧乙烯醚硫酸钠(AES)溶液的物化、表面及应用性能得出:
(1)SAES和AES的黏弹性差别较大,在老化过程中SAES的黏弹性变化较小,而AES的黏弹性变化较大。AES的最终弹性因子和宏观黏度因子显著高于SAES。
(2)SAES和AES的cmc基本一致,但SAES的γcmc略低于AES。在相同浓度下,SAES在气-液界面的吸附扩散速率较快,动态表面活性优于AES。SAES和AES水溶液在低浓度下的吸附过程都属于混合动力控制吸附。
(3)相同浓度下,SAES在石蜡表面的铺展性能优于AES,SAES对帆布片的润湿性、起泡性和稳泡性、乳化性和耐盐耐碱性弱于AES。
