黏土矿物表面羟基介导的分子氧活化机理
2024-03-25牛慧斌黄应平谈云志陈晓婷方艳芬
杜 晴,牛慧斌,徐 艳,张 静,兰 星,黄应平,谈云志,陈晓婷,方艳芬
(1. 三峡大学材料与化工学院,2. 水利与环境学院,3. 土木与建筑学院, 宜昌 443002)
天然层状黏土矿物因其储量丰富且具有独特的硅酸盐层结构而备受关注. 黏土矿物的单位晶胞系由Si-O四面体和Al-(O,OH)八面体所组成,晶层间能吸附数量可变的层间水,且层间具有较高阳离子(Fe3+,Al3+,Ca2+,Mg2+)交换能力[1]. 这些硅酸盐结构衍生的大量Lewis酸、 碱位点对催化转化有机污染物起到了重要作用. 如Fe(III)-蒙脱石能够通过表面Lewis酸位点[Fe(Ⅲ)]向五氯苯酚(PCP)进行单电子转移,生成活泼的PCP*,从而促进了PCP 在土壤中的分配和转化[2]. 又如黏土表面Lewis 碱位点(Si/Al-O-)使四环素(TC)分子中的氨基和酚基脱去质子,促进了水中TC的降解[3]. 因此,作为一种廉价绿色、 具有多活性位点的天然材料,层状黏土矿物已被广泛用于环境污染控制领域.
分子氧(O2)能否被高效活化是衡量催化剂环境友好的关键因素之一. O2活化是指在酶[4]、 金属有机复合物及光等作用下,O2分子在其π*轨道上两个平行的自旋不成对电子发生的电子转移过程[5,6]. 目前,能高效活化O2的催化剂主要有3类: (1) 共价有机框架(COFs)类,通过构建供体-受体(D-A)结构,加速分子内电荷转移,提高对O2的活化能力[7]. 如卟啉COFs中Zn2+的加入促进了单重态激子向三重态激子的转化,Ni2+的引入则促进了激子在光激发下解离到热载流子. 这两种不同的激子行为使O2在可见光照射下可分别活化为和[8]; (2) 单原子催化剂类,通过形成活化势垒更低的活性物种来活化O2,如Fe/MnO2单原子催化剂表面形成的Fe(O=O)Fe 物种能快速活化O2[9]; (3) 金属氧化物类,如NaH2PO2修饰的TiO2纳米粒子,通过氢键相互作用削弱Ti-O 键,从而诱导氧空位的形成,其形成的Ti-O-P共价键调整了氧空位的电子结构,使双电子O2活化的电子快速转移成为可能,从而促进了O2的活化[10]. 以上活化机制均通过催化剂活性中心原子和载体之间的电荷转移来改变O2的电子态、 调控其在催化剂表面的吸附活化能来实现[11]. 但由于受到复杂的合成过程、 金属原子和载体的选择性等多方面的限制,开发出能高效活化O2的催化剂仍是环境与能源领域目前面临的重大挑战[12].
实际上,天然黏土矿物具备活化O2的能力,但活性普遍太低. 如在太阳光诱导的水铁矿表面Fe(Ⅱ/Ⅲ)氧化还原循环过程中,表面Fe(II)会与O2发生界面电子转移(IET)生成了[12,13]还原态含铁黏土矿物可直接活化O2产生羟基自由基(·OH)、 高价铁[Fe(IV)]等氧化物种来降解有机污染物[14]. 与铁矿物不同,黏土类矿物活化O2的能力则主要依赖于电子供体,包括晶格Si-O-Al 和表面Me-OH两类基团[15~17]. Si-O-Al 键中的氧原子被光活化,将电子转移给黏土表面吸附的O2,从而产生和·OH,促进了全氟辛烷磺酰胺向全氟羧酸的转化[15,16]. Ogawa等[17]根据二硝基苯和甲基紫精能够从SiO2中提取电子的报道[18],推测表面Me-OH同样具有电子供体的特性,但至今并未有直接的实验数据能佐证这个推论. 在以往的报道中,黏土表面Me-OH通常以Brönsted酸通过氢键来吸附底物,如蒙脱土在水桥键的协助下其表面Me-OH通过氢键吸附含氮染料罗丹明B[19],又如高岭土通过表面Me-OH与氯霉素的羰基形成氢键来降低氯霉素的水解活化能,促进其催化水解[20]. 但是,表面Me-OH在活化O2过程中,是作为Brönsted酸还是作为电子供体参与仍不清楚,其催化机制有待深入研究.
本文通过不同温度处理调控了红黏土表面Me-OH的位点数,分析其表面Me-OH结构及其活化O2降解TC的能力; 采用原位漫反射傅里叶变换红外光谱法(In-situDRIFTS)和吡啶吸附实验等手段研究了表面Me-OH对O2的活化机制. 通过实验手段证明了红黏土表面Me-OH位点不能直接作为电子供体活化O2,而是作为Brönsted酸来吸附O2. 该研究为高效绿色的黏土类催化剂的开发提供了一定的理论依据.
1 实验部分
1.1 试剂与仪器
四环素(TC)、 异丙醇(IPA)、 苯醌(BQ)、 组氨酸(His)、 荧光反应剂溶液(主要成分: 邻苯二胺)、 无水硫酸钠(Na2SO4)、 氢氧化钠(NaOH)和盐酸(HCl)均为分析纯,国药集团化学试剂有限公司; 二次蒸馏水自制(电导率1.0 µS/cm,Milli-Q HX7000 SD型纯水仪,德国默克集团); R-Clay试样取自桂林市临桂区,其成分列于表S1(见本文支持信息).
E2690 型高效液相色谱仪(HPLC,美国Waters 公司); JSM-7500F 型冷场发射扫描电子显微镜(SEM,日本JEOL 公司); D/max 2500型X射线衍射仪(XRD,日本Rigaku 公司); IS50型傅里叶变换红外光谱仪(FTIR,美国Nicolet公司); F-4600型荧光分光光度计(PL)、 UV-3010型紫外-可见漫反射分光光度计(UV-Vis DRS,日本Hitachi 公司); SCALAB 250Xi 型X 射线光电子能谱(XPS,美国Thermo Fisher Scientific 公司); 1010E 型电化学工作站,美国Interface 公司; XPA-7 型光反应仪器,麦科仪(北京)科技有限公司; JW-BK 112型比表面积和孔径分析仪[BET,JWGB(中国)公司]; HQ30D型溶氧仪,美国哈希公司; Innova型原子力显微镜(AFM)和A300型电子顺磁/自旋共振波谱仪(EPR/ESR,EPR仪器的设置为: 中心磁场强度0.34867 T; 扫场范围0.01 T; 微波频率100 kHz,功率10.02 mW),德国Bruker公司.
1.2 实验过程
1.2.1 热处理 将2 g R-Clay粉末置于坩埚中,在管式炉中于N2气氛围下进行热处理,以20 ℃/min的速率分别升温至300,400和500 ℃,运行3 h,自然冷却后取出,磨碎,所得棕/灰色粉末状固体即为热处理R-Clay粗品,分别记作R-Clay300,R-Clay400和R-Clay500.
1.2.2 样品表征 为了确定样品的晶体结构信息,在2θ为5°~90°的范围内以10°/min的速率获得了物质的XRD谱图. 采用SEM测试了热处理R-Clay样品的形貌和大小.
采用FTIR对样品的特征红外光谱峰进行了表征,原位红外配置Drift 吡啶实验分析样品表面酸位点类型. 通过比表面积和孔径分析仪在-196 ℃和N2气氛围下研究了比表面积(SSA)和孔径分布情况.样品的光吸收特性在250~700 nm 范围内,以BaSO4为参照物,通过在配备积分球的UV-Vis DRS 进行. 根据Tauc方程(下式)计算出样品的禁带宽度分别为2.41,2.28,2.00和2.20 eV.
式中:α为吸收系数;hν(eV)为光子能量;Eg(eV)为禁带宽度;A和n为拟合参数.
1.2.3 电化学实验 在电化学工作站上用玻碳旋转圆盘(GCD)电极为工作电极、 铂丝为对电极与饱和甘汞为参比电极组成的三电极系统进行光电化学测量,测试所用的电解质为100 mL 0.20 mol/L Na2SO4水溶液,测试样品时保证电极表面催化剂的质量一致,并采用几何电流密度计算电流密度,电极几何面积为0.196 cm2.
1.2.4 R-Clay400样品表面羟基位点数的测定 采用氟离子选择性电极法进行测定,将0.05 g样品分散到10-5mol/L的NaF溶液中,搅拌 3 h后测定氟离子浓度,与初始浓度10-5mol/L 的差值再除以其比表面积,即为样品表面羟基位点的数目[21].
1.2.5 TC降解实验 在磁力搅拌下,分别将含有不同温度热处理的20 mg R-Clay和40 mL 24 mg/L TC水溶液加入光反应管中,放入暗箱中,3 h后达到吸附-解吸平衡. 再使用300 W氙气灯提供入射光,加入滤波片,只保留420~780 nm范围内可见光. 通过冷却水循环器将光催化反应器保持在20 ℃,在不同的时间间隔取样. 用0.22 µm针筒式过滤器过滤以去除固体催化剂,采用紫外-可见分光光度计检测上层清液中TC 的残余,最大吸收波长λ=360 nm,最低检出限为0.5 mg/L,线性范围: 0.5~50 mg/L(R2=0.999)(图S1,见本文支持信息). 根据ct/c0~t绘制降解动力学曲线,计算去除率η(%). 为了实验结果的准确性,所有实验进行3次,取平均值. 误差条表示独立实验均值的每个标准差.
式中:c0和ct(mol/L)分别为0和t时刻降解TC的摩尔浓度.
1.2.6 活性物种的检测 采用EPR 对光反应过程中的羟基自由基(·OH)、 超氧自由基和单线态氧(1O2)的生成情况进行了分析. 以水和乙腈分别配置的5,5-二甲基-1-吡咯啉-N-氧化物(DMPO,0.4 mol/L)作为捕获剂,采用EPR测定TC降解过程中的·OH和; 以乙腈配置的2,2,6,6-四甲基哌啶(TEMP,0.2 mol/L)作为捕获剂,测定TC降解过程中的1O2的信号峰; 在室温下(25 ℃)通过波谱仪进行记录,光源为: Quanta-Ray ND: YAG 脉冲(10脉冲每秒)激光系统(λ=532 nm; 10 Hz).
式中:kscavenger和kblank分别为加入捕获剂和不加捕获剂R-Clay400降解TC的速率常数.
1.2.8 原位O2-DRIFTS实验 采用原位漫反射傅里叶变换红外光谱(DRIFTS)(Scheme 1)分析了O2存在下R-Clay400催化剂的Me-OH结构. 以4 cm-1的分辨率采集了1500~4000 cm-1范围内的DRIFTS光谱. 催化剂在25 ℃真空下处理30 min,然后关闭真空泵. 将这些样品在N2气氛围下预处理60 min,采集背景,再将N2气阀门关闭,然后用O2气(流量为0.2×10-3L/min)吹扫90 min,然后关闭O2气阀门,通入N2气吹扫100 min,采用红外光谱在线连续记录.
1.2.9 原位吡啶-DRIFTS实验 以吡啶为探针分子,采用DRIFTS研究了R-Clay400催化剂的表面酸位点类型. 采集过程同上,通过N2气流导入适量吡啶蒸气,并在不同的吸附时间下采集序列. 停止导入吡啶,只通入N2气,采集脱附下的序列. 扫描范围为1300~1800 cm-1,分辨率为4 cm-1.
1.2.10 H2O2的检测 取降解液0.5 mL,用蒸馏水稀释至4倍,再加入50 µL 0.2 mol/L的荧光反应剂溶液,振荡均匀,混合10 min 后,加入1 mL 0.1 mol/LNaOH,用PL 测定样品的荧光强度,荧光光谱仪参数设定: 激发波长300 nm,发射波长400 nm[23].
2 结果与讨论
2.1 热处理R-Clay的结构变化
随着热处理温度的升高,R-Clay 颜色由原始浅黄色变为红棕色(300,400 ℃),最后变为灰褐色(500 ℃)(图S2,见本文支持信息). 用SEM 和AFM 观察了所制备样品的形貌特征,结果显示,R-Clay片层边缘较模糊[图1(A)],厚度在770~1150 nm之间(图S3,见本文支持信息),与其相比,热处理后的R-Clay300,R-Clay400和R-Clay500的厚度分别减少了499,813 和696 nm(图S3),此外,可以看到,R-Clay400和R-Clay500样品中有较多100 nm以内的小片层,这可能是由于热处理后样品的水分损失导致其黏度降低、 层状结构更为明显,且使得大片层转变为更多的小片层,更加有利于表面活性位点的暴露. R-Clay 热重分析结果表明,在258 ℃之前主要脱去吸附水,258~400 ℃之间主要脱去层间水,400 ℃以后则脱去了晶格羟基(图S4,见本文支持信息)[24,25]. 说明R-Clay300和R-Clay400脱去了吸附水和层间水,R-Clay500则脱去了吸附水、 层间水和晶格羟基.

Fig.1 SEM images of R-Clay(A), R-Clay300(B), R-Clay400(C) and R-Clay500(D)
热处理后的R-Clay 随着水分子的脱去导致其接触角也随之增大(40.0°<79.0°<96.5°<119.0°)(图S5,见本文支持信息),随着疏水性增强,其层状结构明显,这与SEM结果相符. 疏水性增强也导致R-Clay 黏度降低,有利于增加比表面积[26]. 由BET 测试结果(表S2,见本文支持信息)可知,与R-Clay 相比,R-Clay300的比表面积显著增加了34.513 m2/g; 但因温度过高,部分结构坍塌,导致R-Clay400和R-Clay500比R-Clay300分别减小10.336和21.156 m2/g. 与此同时,R-Clay表面羟基(-OH)的数量随温度的升高从17 sites/nm2减少至约10 sites/nm2,这主要是由热处理导致(吸附/层间)水及(晶格)羟基失去导致的(图S4).
采用XRD 来分析样品的晶相组成,如图2(A)所示,可见,在R-Clay 和R-Clay300的(001),晶面上分别出现了明显的高岭石特征衍射峰,其中,和晶面对应Fe2O3的特征峰. 随着热处理温度的升高,R-Clay400和R-Clay500对应的高岭石特征衍射峰全部消失,而Fe2O3峰变得尖锐,以上结果说明,通过400 ℃以上热处理,R-Clay 中高岭土结构会消失,而Fe2O3晶型变得更清晰.
采用FTIR 进一步分析了不同温度热处理前后R-Clay 表面官能团的变化. R-Clay 样品在911和1001 cm-1处的强吸收峰分别归属于Si-O 键和Si-O-Si 键的伸缩振动[19][图2(B)]. 与R-Clay 相比,R-Clay300和R-Clay400仍存在着Si-O键和Si-O-Si键的吸收峰,而R-Clay500样品已有部分Si-O键消失.
同样,R-Clay 样品在680 和795 cm-1处的峰归属于Al-O 键的伸缩振动[19],热处理后,R-Clay300和R-Clay400仍存在这两个峰,而R-Clay500中消失,说明R-Clay300和R-Clay400中Al-O 键依然存在,而R-Clay500中Al-O 键全部断裂. 此外,R-Clay 样品在519~527 cm-1处的峰归属于Fe2O3上Fe-O 键的弯曲振动. 以上结果与XRD结果一致,再次证明了高岭石结构的消失,以及Fe2O3的存在.
讨论了表面Me-OH的存在. R-Clay 在3447和3556 cm-1处的峰分别归属于H-OH和Fe-Fe-OH的伸缩振动[19,27],这两个峰在热处理(300~500 ℃)后消失,与热重分析结果一致,经热处理导致了R-Clay层间水和晶格羟基脱失. 值得注意的是,3617,3650 和3695 cm-1处的3 个峰分别归属于晶格羟基(Al-Fe-OH)和表面羟基(Al-Al-OH,Al-Si-OH)的伸缩振动[27~31]. R-Clay500的这3个峰已消失,而R-Clay300和R-Clay400的仍存在,说明这两个样品表面存在着晶格羟基和表面羟基,其中晶格羟基(Al-Fe-OH)主要以八面体位点的Fe(III)存在[27],在蒙脱石框架内,三氯联吡啶钌([Ru(bpy)3]2+)[32]和甲基紫精(MV2+)[33]的激发态会被氧化态Fe(Ⅲ)猝灭,这意味着Clay虽有晶格羟基(Al-Fe-OH),但会因Fe(Ⅲ)的存在猝灭掉电子,故无法活化O2. 为了进一步排除晶格羟基的作用,分析了R-Clay400降解反应前后的XPS谱图. 相对Al(1.12%)和Si(0.52%)的元素损失,Fe元素变化最小(减少了0.07%,表S3,见本文支持信息),证明R-Clay300和R-Clay400表面羟基(Al-Al-OH和Al-Si-OH)是主要活化位点.
2.2 活化O2降解TC
4 个样品对TC 的去除性能结果见图3(A). 在暗反应条件下,R-Clay,R-Clay300,R-Clay400和R-Clay500对TC 的吸附率分别为7.75%,22.41%,38.84%和20.16%. 其中,R-Clay400对TC 的吸附率最大,这可能与R-Clay400表面存在的Al-Al-OH和Al-Si-OH两种羟基形态有关. 在可见光照射条件下,与R-Clay相比,热处理后的3个样品对TC的降解能力均有提高. 其中,R-Clay400显示出最佳降解能力,在6 h 内降解效率为86.36%. 通过一级动力学拟合数据显示[图3(B)],R-Clay400的速率常数为0.3380 h-1,分别为R-Clay(0.1228 h-1),R-Clay300(0.2168 h-1)和R-Clay500(0.2577 h-1)的2.75,1.56和1.31倍.说明热处理调控了R-Clay 上的Me-OH 位点数(表S2),使含有Al-Al-OH 和Al-Si-OH 两种羟基的R-Clay400具有活化O2的能力.
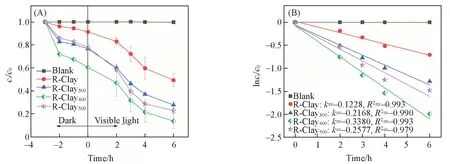
Fig.3 Degradation kinetics of TC(A) and first order dynamics fitting diagram(B) of samplesc0(TC)=24 mg/L,m(catalyst)=20 mg,pH=5.6±0.2,t=25 ℃.
图4显示了不同pH、 不同催化剂用量和不同底物浓度等因素对R-Clay400降解TC的影响. 在pH=5,7 和9 的条件下,TC 的降解率分别为64.15%,75.37%和83.13%,反应速率常数分别为0.156,0.230和0.234 h-1[图4(A)和(A')]. 说明TC在碱性条件下更易发生降解,其原因在于碱性条件下TC的摩尔吸光系数更高,更有利于对可见光的吸收[3],促进了TC的降解.
从图4(B)和(B')可见,随着催化剂用量的增加(10,20 mg),TC 的降解率也随之增大(80.88%,89.26%). 但当催化剂用量增加到30 mg时,其降解率(93.80%)与20 mg相差不大,这可能是因为过多催化剂的加入,影响了TC对可见光的吸收[3],导致其降解率没有随催化剂用量的增加而呈现明显的递增趋势.
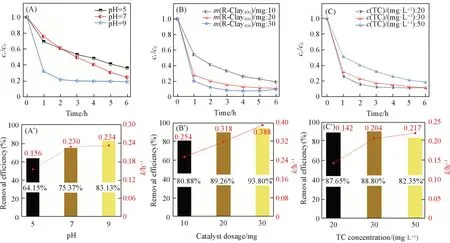
Fig.4 Removal of TC in the presence of R-Clay400 at different pH(A, A'), with dosages catalysts(B, B'),TC concentrations(C, C')
图4(C)和(C')显示了TC浓度对其降解效率的影响,可见,随着TC浓度的增加(20,30,50 mg/L),其降解率(87.65%,88.80%,82.35%)减小了5.3%,这说明TC浓度对其降解效率影响不大. 图S6(见本文支持信息)显示了不同光源对TC降解速率的影响,结果表明,黑暗条件下TC几乎不能被降解,而可见光照射下TC的降解率达到了86.36%. 这说明R-Clay400能够被可见光激发,活化O2产生ROS,从而降解TC. 此外,R-Clay400在6 h内对TC的矿化率达到了40%(图S7,见本文支持信息),表明R-Clay400具有一定矿化TC的能力.
为了进一步证实R-Clay400确实能够活化O2,进行了无氧TC 的降解实验(图5). 可以看到,在无氧(Ar)条件下,R-Clay400对TC的降解完全被抑制,说明TC 的降解依赖于O2的存在. 在通入O2气、 空气和氩气条件下,R-Clay400/TC 体系中溶解氧含量按R-Clay400-O2(21.00 mg/L)>R-Clay400-Air(9.90 mg/L)>R-Clay400-Ar(1.61 mg/L)降低(表S4,见本文支持信息),但是体系中TC 的吸附量反而依次增加,即R-Clay400-Ar 的TC 吸附量最高(24.26 mg/g),说明O2和TC对R-Clay400表面吸附位点存在竞争. 此外,随着溶氧量的增加(1.61 mg/L→9.90 mg/L),R-Clay400对TC的降解率从无氧条件下的10.45%升高到89.87%,说明R-Clay400主要是通过活化O2产生的ROS来降解TC的.
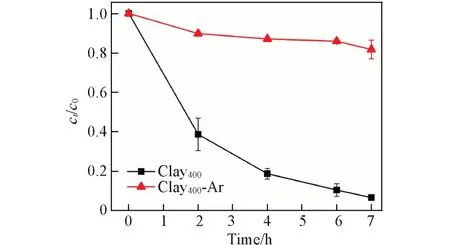
Fig.5 Removal of TC in the presence of RClay400 in different atmospheres
通过O1s的XPS 谱图来观察R-Clay 表面含氧基团的含量随热处理温度的变化. 529.99,531.87 和532.75 eV 附近的峰分别归属于表面晶格氧物种(OL)、 表面羟基物种(OOH)和吸附氧物种(Oads)[图6(A)][34,35].
由图6(B)和(D)可见,R-Clay300和R-Clay500的OOH,Oads的百分比与R-Clay 相当. 由图6(C)可见,R-Clay400表面的OOH占比(23.4%),比原始R-Clay(57.1%)减少了33.7%,而Oads占比(72.4%)比R-Clay(34.5%)增加了37.9%,这暗示着R-Clay400表面OOH的下降,与其表面O2吸附含量有关. 当R-Clay400表面Me-OH 占比最少时,其表面吸附的O2含量最高,表明O2活化与表面Me-OH 位点数有关. 这说明在热处理中R-Clay400表面Me-OH已被O2占据而导致其含量最少,吸附O2量最多.

Fig.6 O1s XPS spectra of R-Clay(A), R-Clay300(B), R-Clay400(C) and R-Clay500(D)
2.3 O2活化机制
2.3.1 ROS 的生成 为了分析R-Clay400在降解TC 过程中产生的ROS 类型,采用EPR 技术和捕获实验进行了测试. 用DMPO和TEMP探针分子来监测R-Clay400体系光解过程中的活性自由基. 图7(A)显示了DMPO-·OH,和在R-Clay400和可见光的辐照下产生的自旋加合物的EPR光谱. 可见,和具有明显的信号峰,而DMPO-·OH未监测到信号峰. 表明R-Clay400体系中产生的主要活性自由基是和另外,在捕获剂组氨酸(His,捕获1O2)和对苯醌(BQ,捕获)存在的条件下,R-Clay400对TC的降解速率出现不同程度的降低[图7(B)],证实R-Clay400体系产生的活性物种有和而异丙醇(IPA,捕获·OH)的加入对TC的降解完全没有影响[图7(B)],再次验证了体系中不存在·OH. 此外,通过分析ROS对TC降解的相对贡献(图S8,见本文支持信息),发现和的相对贡献率分别为64.28%和35.72%. 进一步说明活化O2生成的和是造成TC降解的主要ROS.
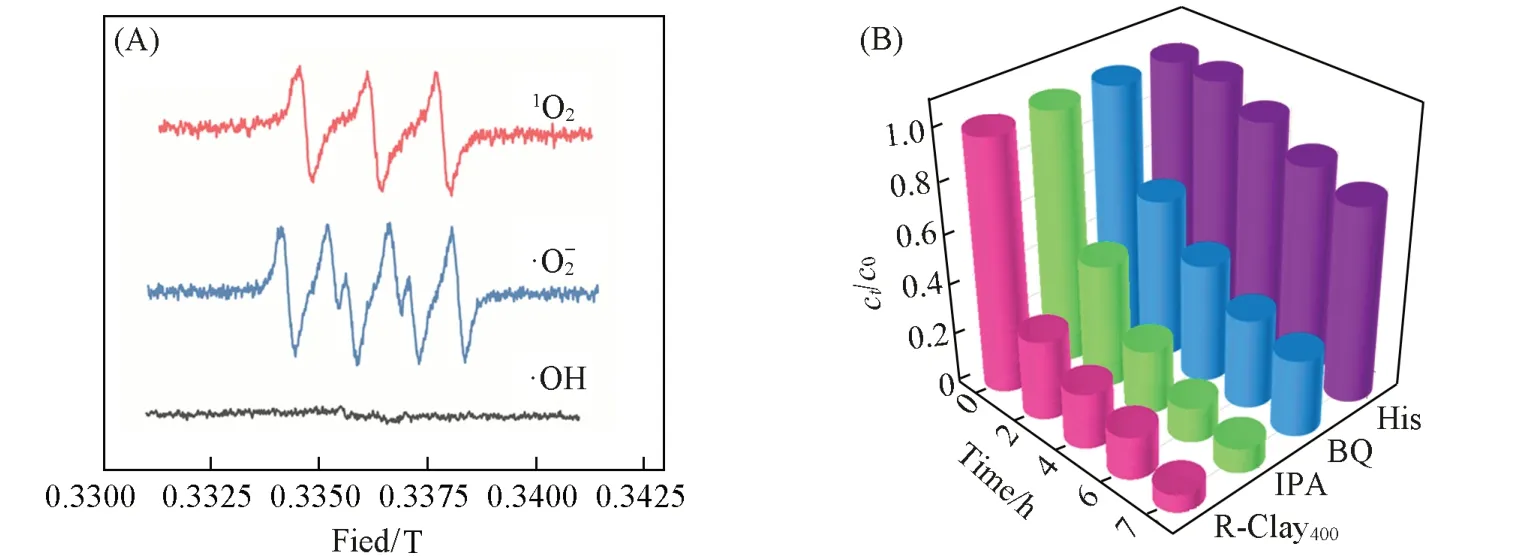
Fig.7 EPR spectra of R-Clay400 in light(A) and capture experiment(B) of samples
2.3.2 表面羟基Me-OH协同O2活化机制 为了揭示O2活化机理,进行了原位O2吸附的DRIFTS实验.图8(A)显示了R-Clay400样品在1500~4000 cm-1范围内的原位O2-DRIFTS光谱. 在3622和3697 cm-1处出现的负峰归因于Me-OH 的伸缩振动[27~31],这是由于R-Clay400上Me-OH 与O2发生了化学吸附,使得Me-O-H键电子云密度降低/断键,实现Me-OH与O2之间电子云密度的转移. 同时,1648 cm-1处的新峰归属为O2的O与Me-OH的H的弯曲振动,这是由于电子云密度的转移导致它们成键. 经N2气吹扫,发现Me-OH与O2之间位于3622,3697 cm-1处的负吸收峰和1648 cm-1处的正吸收峰基本消失(图S9,见本文支持信息),进一步表明,Me-OH与O2之间生成了氢键. 由R-Clay400反应前后的O1sXPS谱图发现,反应后R-Clay400上Oads的结合能(531.9 eV)比未处理R-Clay(532.25 eV)蓝移了0.35 eV[图8(B)],而OOH的结合能(531.49 eV)比反应前的(531.20 eV)红移了0.29 eV,证明Me-OH和O2之间的确形成了氢键.
为了进一步探究氢键的类型,进行了R-Clay400样品的原位吡啶吸附的DRIFTS 实验. 图8(C)显示了R-Clay400样品在1300~1800 cm-1范围内的原位吡啶-DRIFTS 光谱. 位于1439,1482 和1580 cm-1处的吸收峰归因于吡啶分子骨架上C-H 和平面内环的形变振动. 位于1540 cm-1处的吸收峰归因于吸附在Me-OH(吡啶-B)上的吡啶[36],这证明R-Clay400表面存在明显的Brönsted酸位点. 同样,原位吡啶脱附的DRIFTS 光谱(图S10,见本文支持信息)中存在1540 cm-1处的吸收峰,再次证明了强Brönsted 酸位点的存在. 因此,R-Clay400表面Me-OH 并非Ogawa 等[17]推测的能够作为电子供体活化O2,而是通过Brönsted 酸位点以氢键的形式吸附了O2,此结论通过实验得到了证实. 由此可见,R-Clay400表面Brönsted酸介导的O2吸附对其活化作用尤为重要.
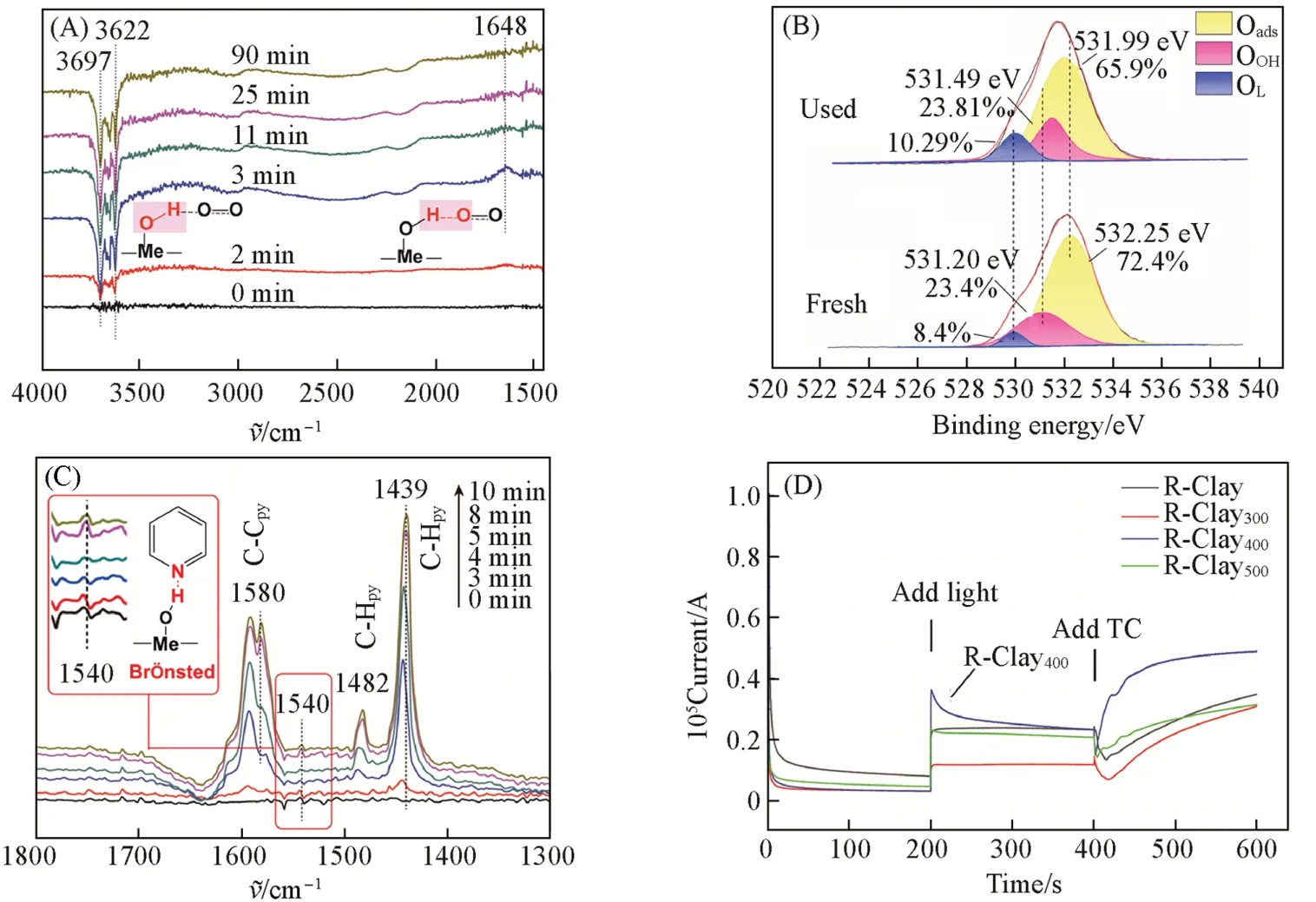
Fig.8 In situ O2-DRIFTS spectra of R-Clay400 samples(A), O1s XPS spectra of R-Clay400(B), in situ pyridine-DRIFTS spectra of R-Clay400 samples(C) and current response under different conditions of samples(D)
在可见光光照作用下,O2需要得到光生电子才能发生活化,生成·O2-[37]. 能提供光生电子的物质只可能是能吸收可见光的R-Clay400或底物TC. 为了分析光生电子的来源,测试了样品的UV-Vis DRS 光谱,发现以Fe2O3为主要组成的R-Clay400能吸收675 nm以下的可见光(图S11,见本文支持信息)[38]. 通过光致发光、 产氢性能测试和电化学等实验发现,与其它样品相比,R-Clay400的光致发光强度最低、 产氢效率最高、 光电流响应最强、 电化学阻抗(EIS)最小(图S12,见本文支持信息),证明在可见光激发下,R-Clay400最易发生光生电子-空穴分离,分离的光生电子被表面吸附的O2捕获,产生[13]). 结合DRIFTS实验,进一步表明R-Clay400的确能够作为电子供体活化O2,且起活化作用的主要是Si-O-Al的O原子而非Me-OH的O[17]. 此外,为了判断TC在可见光激发下能否也产生光生电子[39],测试了在不同操作条件下R-Clay400的电流响应图[图8(D)]. 发现单独R-Clay400的电流维持在较低的值,200 s后增加可见光照,R-Clay400的电流响应增强,说明了R-Clay400在可见光的激发下电子转移速率加快,再次证明了R-Clay400作为电子供体可活化O2. 400 s后再添加TC,发现R-Clay400对电流的响应强度显著增加,证明了TC和R-Clay400之间存在着电子转移[40]. 另外,加入TC后的R-Clay400的EIS奈奎斯特弧半径明显小于R-Clay400的(图S13,见本文支持信息). 通常,EIS奈奎斯特弧半圆的半径越小,电阻越低,界面电荷传输效率越高[41]. 以上结果都证明TC和R-Clay400之间确实存在电子转移,即TC也能作为电子供体来活化O2. 此外,瞬时电流实验发现(图S14,见本文支持信息),通入氧气,R-Clay400的瞬态电流发生小幅度的变化,再加入TC,瞬态电流再次改变,再次证明R-Clay400表面Me-OH能与富电子的O2和TC成键,光照加速了它们之间的电子转移.
上述实验证明了R-Clay400具有活化O2的能力,但是否依赖于Me-OH对O2的吸附还需要更直观的证据. 为此,测试了随着降解反应的进行,R-Clay400表面Me-OH位点数的变化(表S5,见本文支持信息),发现光反应在0~6 h 内活性位点逐渐减少: Light 0(15.00 sites/nm2)>Light 4(14.53 sites/nm2)>Light 6(14.48 sites/nm2),说明在TC 光催化降解过程中Me-OH 确实有参与. 但因吸附的O2被不断活化而消耗,无法定量地测定降解过程中吸附与活化速率. 根据原位DRIFTS实验可知,R-Clay400表面Me-OH通过氢键能直接吸附固定O2,将有助于光生电子的转移. 此外,有报道显示,能被H2O氧化为,同时生成H2O2和OH-[42]. H2O2的产生将有助于证实该反应发生. 随着反应时间的延长,发现H2O2的荧光强度越强,说明了确实被氧化并生成了H2O2(图S15,见本文支持信息). 与反应前pH 值(pH=5.6±0.2)相比,反应后pH值升高(pH=6.21±0.2),也说明被氧化为并释放出OH-.
综上可知,R-Clay400通过界面电子转移(IET)[13]的方式活化了O2,推测其可能的反应历程见式(4)~式(9). 首先,R-Clay400表面Me-OH作为 Brönsted酸位点通过形成氢键吸附O2. 同时,在可见光的照射下,R-Clay400和TC作为电子供体产生的光生电子转移给相邻吸附态O2,生成了,接着,与H2O反应生成和H2O2. R-Clay400表面Me-OH活化分子氧机制见Scheme 2.

Scheme 2 Mechanism diagram of molecular oxygen activation by the Me-OH sites on the surface of R-Clay400
3 结 论
通过热处理调节R-Clay表面Me-OH的位点数,采用SEM,XRD,XPS,FTIR和DRIFTS气固原位红外对其物理性质进行表征. 结果表明,R-Clay400表面Me-OH以Brönsted酸形式存在,因吸附了大量O2,导致其表面Me-OH含量最低. R-Clay400对TC具有最高去除率(86.36%,k=0.3380 h-1)和良好矿化效果(40%). 在可见光照射下,R-Clay400的Me-OH作为Brönsted酸位点通过氢键吸附O2,吸附态的O2得到从R-Clay400和TC转移过来的光生电子生成又进一步被氧化为,实现了TC的降解. 本研究进一步阐明了黏土矿物对O2活化机理,促进了矿物基材料相关研究领域的发展.
支持信息见http: //www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20230422.
