基于原子特性知识增强的分子毒性预测方法
2024-03-25方舒言侯阿龙秦欢欢
方舒言,刘 宇,2*,侯阿龙,秦欢欢,刘 嵩,4
(1.武汉科技大学 计算机科学与技术学院,湖北 武汉 430072;2.湖北省智能信息处理与实时工业系统重点实验室,湖北 武汉 430072;3.武汉科技大学 医学院,湖北 武汉 430072;4.湖北省职业危害识别与控制湖北省重点实验室,湖北 武汉 430072)
0 引 言
新药研发存在周期长、费用高和成功率低等特点。人工智能技术是近些年来的热点技术之一,在很多领域都有非常广泛的应用,多种人工智能方法已经成功应用于药物的发现过程[1]。当前化学分子毒性预测方法主要从分子结构的序列表示中学习特征,如SMILES[2](Simplified Molecular Input Line Entry System)。SMILES通过对分子图的生成树实施深度优先的前序遍历,为每个原子、键、树遍历决策和断环生成符号,定义了分子的字符串表示,所得到的字符串对应于分子图生成树的序列化。因比其他表示结构的方法(包括图)更紧凑,SMILES已被广泛用于分子毒性预测。
目前,以Chemberta[3]为代表的分子毒性预测方法借鉴了自然语言处理技术的思想,直接使用大量无标注的SMILES作为语料库来学习分子表征,并使用学习到的分子表示用于毒性预测。然而SMILES语法复杂且限制性强,常规字符集上的大多数序列不能明确定义分子。
由于上述限制,基于SMILES的深度学习模型只能将学习重点放在分子串的语法上,导致现有的毒性预测方法仅损失了分子中的原子特性等知识。针对上述问题,以Mol-BERT[4]为代表的方法选择了使用摩根指纹标识符(Morgan Fingerprint Identifier,MFI)序列作为输入来进行模型的训练,并在MFI融入了原子特性的知识(如原子序数、电荷量、原子的度等)。摩根指纹(Morgan Fingerprints)是一种使用哈希算法生成的圆形分子指纹[5-6]。其中,原子的MFI是对原子及其属性(如原子序数、电荷量等)进行哈希得到的一个定长整数,而子结构MFI则是对分子中某原子MFI及以该原子为中心一定半径范围内的其他原子的MFI进行联合哈希得到的定长整数。根据设置的指纹半径的不同,可以得到蕴含不同知识的MFI,比如以某原子为中心且半径为0生成的MFI仅包含该原子的知识,以某原子为中心且半径为1生成的MFI,则蕴含该原子及相邻原子子结构知识。
根据上述对摩根指纹及MFI的说明可知,Mol-BERT虽在一定程度上融入了原子特性的知识,但是仍然存在局限。比如,Mol-BERT的输入MFI序列中未显式引入氢原子的知识,而且对于MFI序列的半径信息Mol-BERT方法并没有进行显式的编码。
针对目前基于分子序列的毒性预测方法均未考虑氢原子和摩根指纹半径等领域知识的问题,该文提出了一种基于原子特性知识增强的分子毒性预测方法。一方面,改进的分子毒性预测方法考虑了分子中所有氢原子,输入的MFI序列显式引入了氢原子的MFI和以氢原子为中心的子结构的MFI。在考虑了分子中所有的氢原子之后,子结构MFI所代表的子结构粒度更小,模型的MFI字典大小变为引入氢原子之前的约一半。这是由于在同样的半径限制下,使得原本一些大而稀有的子结构因为氢原子的引入将被转换成小而常见的子结构,如图1(b)所示。该现象类似于自然语言处理领域的WordPiece[7],它将单词拆成词根和词缀,一些生僻词被拆成常见词根和词缀的组合,能够减小自然语言处理模型的词表大小。另一方面,受SongNet[8]的启发,为了让模型学习MFI序列时可以区分不同半径的MFI,提出使用摩根指纹半径的知识扩展嵌入层的方法,使模型学习到的原子MFI和子结构MFI有所区分。图1(a)~(c)描述了氢原子和摩根指纹半径知识增强MFI序列的过程及增强后MFI序列与原本MFI序列的区别。该文的主要贡献包括3个方面:

(a)原本的MFI序列
(1)提出生成分子MFI序列时显式地引入氢原子的方法,使得分子毒性预测模型可以学习到氢原子信息,同时减少了MFI字典的大小和模型的参数量;
(2)改进的分子毒性预测方法,额外考虑了各原子及相邻原子构成的子结构的知识,并在模型的嵌入层强化了摩根指纹半径知识;
(3)为了验证改进方法的有效性,利用Pubchem[9]分子库中的1 000万分子预训练了基于原子特性增强的分子毒性预测模型(Hydrogen and Radius enhanced Toxicity Prediction model,HRToxPred),并在主流的毒性预测数据集Tox21[10]上进行了验证。
1 相关工作
在基于深度学习的分子毒性预测领域中,主流方法直接在分子的SMILES字符串上训练模型来预测分子毒性。表1总结了基于分子序列的毒性预测模型的相关工作。FP2VEC[11]和Mol2vec[12]使用了自然语言处理领域的Word2vec[13]思想。其中,Mol2vec学习的是MFI的低维向量表示,然后输入学习到的MFI向量表示至下游网络中预测分子毒性,而FP2VEC学习的是比特分子指纹比特串的低维向量表示。然而,在自然语言处理领域中Word2vec的词和向量是一对一的关系,所以无法解决多义词的问题。在化学领域,同样存在类似“一词多义”的场景,例如相同的原子所处的环境不同其作用也不同,所以FP2VEC,Mol2vec存在着类似的问题。Chemberta和Mol-BERT使用了基于Transformer的掩码语言模型。其中,Chemberta设计了一种SMILES的分词方法,使用Roberta[14]的预训练策略练了基于SMILES的分子毒性预测模型,然而SMILES的分词方法需要化学领域的专家来设计,需要消耗大量人力。Mol-BERT使用分子的MFI序列作为语料库,使用BERT[15]的MLM(Masked Language Model)策略预训练了基于MFI序列的分子毒性预测模型。相比于Chemberta,Mol-BERT所使用的MFI序列不需要人工分词。然而Mol-BERT使用的MFI序列未显式引入氢原子且序列中原子指纹与子结构指纹混杂在一起,使模型学习到的分子表示并未区分原子指纹与子结构指纹。针对上述方法存在的问题,该文提出生成MFI序列时显式引入氢原子并对半径信息进行显式编码的知识增强方法。

表1 相关工作总结
2 方 法
2.1 摩根指纹标识符序列
为了便于解释显式引入氢原子后MFI的计算方法,现给出以下定义:
定义1 原子邻域(Atom Neighborhood):给定一个分子图mg=(A,B),A为该分子中的所有原子的集合,B为该分子中所有的化学键的集合。对于一个原子a∈A,给定半径r,以a为中心,r为半径的原子邻域定义为nbr(mg,a,r)={[a,at,b]|a∈A,at∈A,b∈B,a≠at,dist(a,at)≤r,b=
将原子邻域的半径设为1,计算MFI时需考虑到两种不同规模的原子邻域nbr(mg,a,0)和nbr(mg,a,1)。如图2所示,深色节点对应nbr(mg,a,0),而由深色节点a和浅色节点at组成的子结构对应nbr(mg,a,1)。由此,原子的MFI和子结构的MFI将由其对应的原子邻域特性经过哈希得到。

图2 原子邻域、摩根指纹、摩根指纹标识符之间的关系
定义2 摩根指纹[5-6](Morgan Fingerprint):摩根指纹是一个稀疏的比特串。一个分子可以通过散列分子中每个原子的邻域信息得到一个索引,该索引位置的比特位被置为1。
定义3 摩根指纹标识符(Morgan Fingerprint Identifier,MFI):摩根指纹标识符是一个固定长度的整数,用来表示原子邻域的特征。通常情况下,一个原子邻域的摩根指纹标识符可以对该原子邻域的属性(原子序数、电荷量等)进行哈希来获得。记为mfinbr(mg,a,r)=Hash(Attrs(nbr(mg,a,r))),Hash(·)为哈希函数,Attrs(·)表示获取原子邻域的属性。
原子邻域、摩根指纹和摩根指纹标识符之间的关系如图2所示。对于一个分子经过算法1的计算,可以获得该分子的MFI字典。字典的键为MFI,字典的值为由该MFI对应原子邻域中心原子的原子索引和原子邻域半径组成的元组[a.Index,r]。该分子的摩根指纹则是一个长的比特串,其中索引为MFI字典中键值的比特位被置为1,如图2箭头指示。
定义4 原子的摩根指纹标识符:给定一个分子mg=(A,B)和一个原子a∈A,该原子的摩根指纹标识符可以通过哈希原子邻域nbr(mg,a,0)的属性得到。
算法1
输入:分子图mg=(A,B),半径r
输出:存储该分子MFI的字典D
Initialization;
/*初始化一个容器存所有的mfi,偏移量为原子索引*/
Vector
fora∈Ado
/*初始化一个容器存Attrs(nbr(mg,a,0))*/
Vector
components.pushback(a.AtomicNum);//原子序数
components.pushback(a.TotalDegree);//原子的度
ifa≠Hydrogen then
//非氢原子连接的氢原子数
components.pushback(a.TotalNumHs);
end
components.pushback(a.FormalCharge);//原子的电荷量
invars.pushback(boost::hash(components));
end
/*将mfinbr(mg,a,0)的信息存入字典*/
fora∈Ado
D[invars[a.Index]].pushback(Tuple(a.Index,0));
end
/*初始化容器存mfinbr(mg,a,1)*/
Vector
fora∈Ado
/*获取从a出发的化学键*/
bonds = getChemicalBonds(a);
for
ifa≠Hydrogen then
nbrs.pushback(Tuple(
invars[at.Index]));
end
end
invar = 0;
boost::hashcom bine(invar, invars[a.Index]);
for nbr ∈ nbrs do
boost::hashcombine(invar, nbr);
end
/*将mfinbr(mg,a,1)的信息存入字典*/
D[invar].pushback(Tuple(a.Index, 1));
end
该文考虑的原子邻域nbr(mg,a,0)的属性包含原子序数、电荷量、非氢原子连接的氢原子数、原子在分子图中的度。算法1的第3~18行具体描述了该计算摩根原子MFI的过程。
定义5 子结构的摩根指纹标识符:给定一个分子mg=(A,B)和一个原子a∈A,以a为中心的子结构的摩根指纹标识符可以通过哈希原子邻域nbr(mg,a,1)的属性得到。
该文考虑的原子邻域nbr(mg,a,1)的属性包含该原子邻域中化学键b的类型、该原子邻域中原子a和at的摩根指纹。算法1的19~37行具体描述了计算过程。
定义6 摩根指纹标识符序列:摩根指纹标识符序列是由一个分子中所有的原子邻域的摩根指纹标识符组成的,摩根指纹标识符的顺序按照SMILES定义的原子顺序排列。如果对于分子中某原子a有多个摩根指纹标识符,则按照r由小到大进行排序。
根据以上定义,算法1描述了计算最大半径设置为1时的分子的MFI的过程。
2.2 数据预处理
数据预处理阶段,因为同一个分子可以有多种不同的SMILES形式,所以首先要对分子SMILES进行标准化。SMILES之所以出现不同是因为可以随意修改原子的“出场顺序”以得到不同的SMILES。该文使用Rdkit[16-17]实现了标准化过程,并根据分子化合价态平衡原理补全SMILES中的氢原子。最后使用算法1计算并排序生产分子的MFI序列。
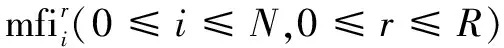
根据上述方法,该文选择Pubchem分子库中1 000万分子的SMILES,并使用算法1分子的MFI并排序形成MFI序列。整个过程中有关分子的处理都使用Rdkit分子处理工具实现。统计1 000万分子的是否显式引入氢原子的MFI序列,得到不同MFI序列的MFI字典信息,如表2所示。

表2 是否引入氢的MFI字典大小比较
显式引入氢原子MFI之后,分子中的某些相对较大的子结构标识符可以分解为一些相对较小的子结构的组合,例如图1中以该羟基(-OH)上氧原子为中心半径为1的子结构会被拆分成以羟基中氢原子为中心半径为1的羟基结构和以氧原子为中心半径为1的结构。这类似于自然语言处理领域把单词拆成词根与词缀的方法。因此,引入显式表示氢原子之后,生成的MFI字典大小更小且MFI对应的原子邻域粒度更细。该文尝试扩展算法1的最大半径限制,当最大半径达到2时,即便引入了氢原子一定程度上减小了MFI字典的规模,然而最大半径限制为2时MFI字典的大小超过了100万,因此该文仅考虑最大半径限制为1的情况。
2.3 模型架构
HRToxPred包含Embedding模块、Transformer模块和下游任务模块,如图3所示。图中M为输入的MFI,F为MFI嵌入(Token嵌入),P为位置编码(Position嵌入),R为半径嵌入(Radius嵌入)。因为生成的每个MFI序列代表一个分子,而每个分子都是单独的个体,无需考虑两个分子之间的关系,所以文中的预训练去掉了BERT模型预训练策略中“下一句预测”(Next Sentence Prediction,NSP)预训练任务,输入样本变为单个MFI序列,因此BERT模型原本用来区分两个句子的Segment编码失去了意义。该文修改了BERT的Embedding模块,将Segment嵌入改为摩根指纹半径嵌入来区分MFI的类别,最终Embedding模块包含半径嵌入、位置嵌入和MFI编码嵌入。
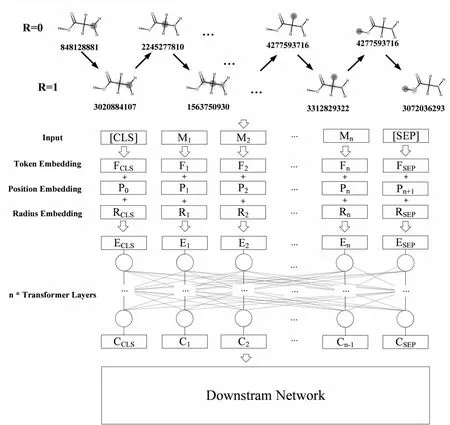
图3 模型架构示意图

经过上述步骤输入数据就处理完成,接下来进行嵌入。对于输入特征,首先要获得MFI编码、半径编码、位置编码的嵌入,通过把所有嵌入向量相加的方式,公式如下:
Ht=Emt+Ept+Ert
(1)
式中,t为序列中MFI的索引,Emt,Ept,Ert分别为MFI嵌入、位置嵌入和半径嵌入,即通过半径嵌入Ert区分了原子MFI和子结构MFI。处理完输入数据和嵌入之后,HRToxPred使用自注意力机制进行学习。
qt=WQHt
(2)
kt=WKHt
(3)
vt=WVHt
(4)
ct=self-att(qt,kt,vt)
(5)
式中,WQ,WK,WV为参数矩阵,self-att(·)为注意力函数,Ct为该注意力头输出。根据上述方法将得到的所有注意力头的Ct向量进行拼接,然后与参数矩阵W求积得到输出c,再将c输入到激活函数中得到对应的输出o。在下游任务中只需将o输入到下游任务的分类器网络中即可。式1~7描述了模型的计算过程。
c=Concat(ct)W
(6)
o=max(0,ZW1+b1)W2+b2
(7)
3 实验与分析
3.1 基准模型
选取了当前被广泛使用的基于分子序列的毒性预测模型Chemberta和Mol-BERT作为对比模型。其中,Chemberta基于Roberta模型实现。预训练时,使用1 000万分子的SMILES作为语料库,随机遮蔽了每个输入字符串中15%的Token。在学习恢复Mask Token时,该模型形成了一个化学空间的分子结构表示。Mol-BERT使用未显式引入氢原子MFI和摩根指纹半径特征的MFI序列作为输入。Mol-BERT同样使用了1 000万分子的MFI序列,并利用了BERT的架构及预训练策略进行了预训练,最后将预训练好的模型进行下游的毒性预测任务。
3.2 预训练
HRToxPred与主流的自然语言处理预训练语言模型的方式不同,去掉了NSP预训练任务,仅留MLM任务。具体来说,该文使用bert-base的模型设置,并将分子MFI序列中随机15%的标识符屏蔽为[MASK]。此外,预训练使用了动态掩码,即同一个MFI序列会产生不同的掩码序列,这样可以重复使用预训练样本,而且可以防止过拟合。
3.3 微 调
选择主流毒性预测数据集Tox21的12个分类任务进行微调。数据集详情见表3。

表3 Tox21各任务细节
3.4 评估标准
由于该文选取的数据集正负样本不平衡,而精度、准确率、召回率等指标受正负样本不平衡的影响,不能客观反映出模型的性能,且在分子毒性预测中区分正负样本的概率阈值并不确定,所以选择AUC[18](Area Under Curve)作为评测指标可避免以上因素带来的影响。
AUC被定义为ROC(Receiver Operating Characteristic)曲线下的面积。ROC曲线,又称接受者操作特征曲线。二分类任务分类阈值的设定不同会得出不同的真阳率(True Positive Rate,TPR)和假阳率(False Positive Rate,FPR),将同一模型每个阈值的(FPR,TPR)坐标都画在ROC空间里,就成为特定模型的ROC曲线。AUC计算公式如下:
(8)
式中,P为正样本数量,N为负样本数量,pi为正样本预测得分,nj为负样本预测得分。
3.5 实验设置
HRToxPred包含Embedding模块、Transformer模块和下游任务模块,模型的实现框架使用UER-py[19]。
Embedding模块包含Token嵌入、Position嵌入和半径嵌入。HRToxPred设置的输入序列长度与BERT保持一致均为512个Token。
Transformer模块选择bert-base结构,bert-base包含12个Transformer层和12个自注意力头,隐藏层维度为768。
下游任务模块的微调网络使用UER-py框架中默认的网络,即在预训练完成的模型后接2层全连接层。对于每个数据集,按照80/10/10的比例随机划分为训练集/验证集/测试集,并进行了32个epoch的微调,具体参数见表4。

表4 模型参数设置
3.6 消融实验分析
为了进一步验证HRToxPred中引入的摩根指纹半径特征及考虑氢原子的作用,进行了消融实验,即用Pubchem中的1 000万分子预训练如下四个模型:
•Base:输入MFI序列不显式引入氢原子及其子结构MFI,嵌入层没有额外的摩根指纹半径嵌入;
•AddHS:输入MFI序列不显式引入氢原子及其子结构MFI,嵌入层没有额外的摩根指纹半径嵌入;
•AddRadius:输入MFI序列不显式引入氢原子及其子结构MFI,嵌入层有额外的摩根指纹半径嵌入;
•HRToxPred:MFI序列显式引入氢原子及其子结构MFI,嵌入层有额外的摩根指纹半径嵌入。
然后将上述四个模型在Tox21上微调。如图4所示,使用显式引入氢原子MFI的模型预训练收敛得更快,主要原因是HRToxPred考虑了氢原子及其子结构MFI造成的词表减小。另外,从表5中Tox21不同通道上的上的AUC分数对比来看,仅使用考虑了氢原子MFI序列作为输入(AddHs)和仅引入摩根指纹半径嵌入(AddRadius)都可以提升模型的分类能力,而两者的组合对模型分类能力的提升更显著。HRToxPred相比于Base模型,在sr_p53,nr_ahr,nr_er,nr_ar,sr_are,nr_ar_lbd,nr_er_lbd,sr_hse等任务上取得了显著的提升。具体而言,sr_p53任务提升了4.5%,nr_ahr任务提升了1.7%,nr_er任务提升了3.1%,nr_ar任务提升了7.5%,sr_are任务提升了4.8%,nr_ar_lbd任务提升了2.6%,nr_er_lbd任务提升了1.5%,sr_hse提升了3.3%。而在nr_aromatase,nr_ppar_gamma,sr_atad5,sr_mmp等任务上的表现不如预期。
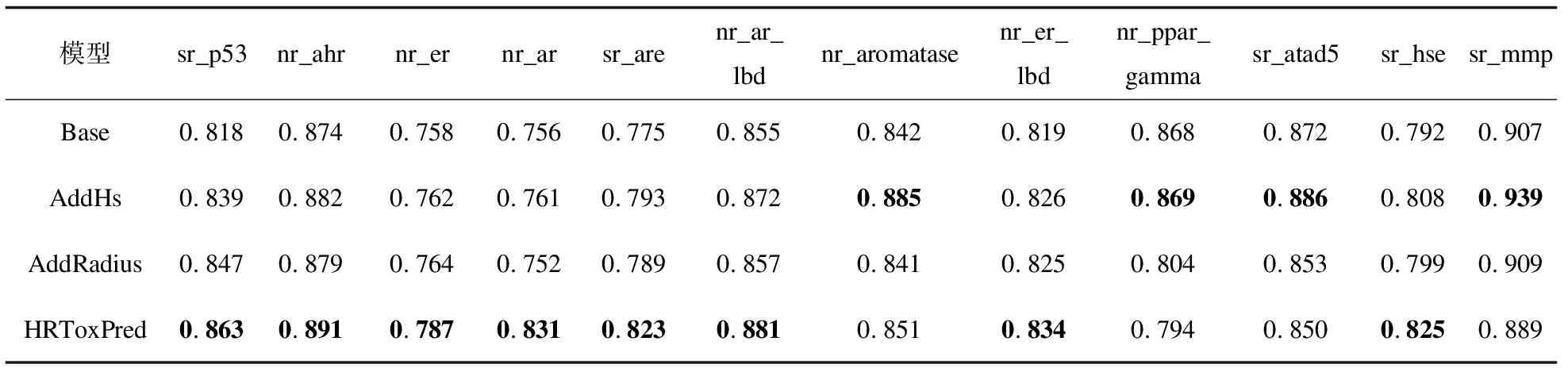
表5 消融实验结果比较(ROC-AUC↑)
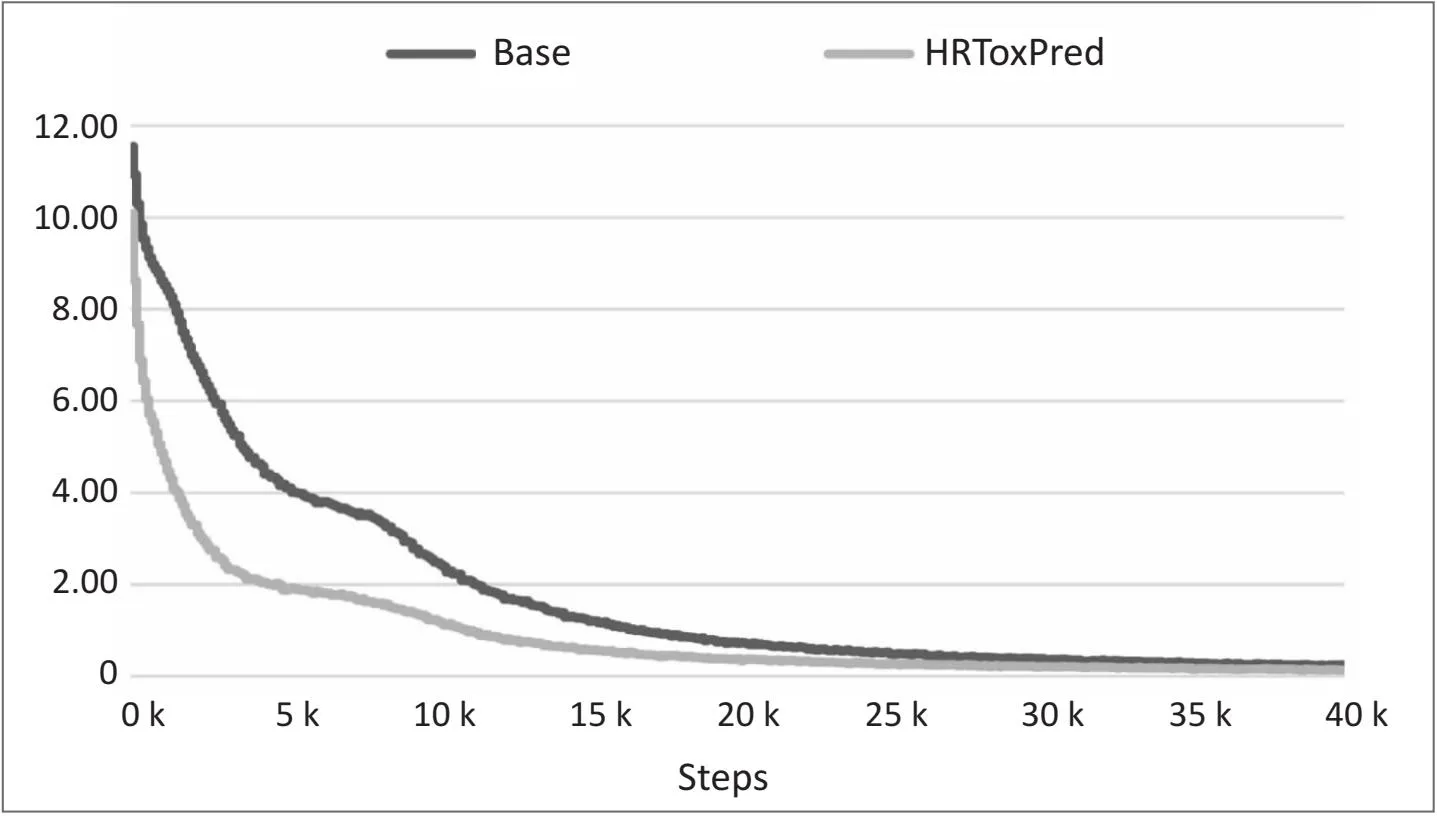
图4 Base和HRToxPred预训练Loss曲线
3.7 对比实验分析
为了检验HRToxPred的有效性,将HRToxPred与Chemberta和Mol-BERT模型进行了比较。表6报告了各个模型Tox21数据集12个通道上的AUC分数。HRToxPred在sr_p53,nr_ahr,nr_er,nr_ar,sr_are,nr_ar_lbd,nr_aromatase,nr_er_lbd,sr_hse等任务上取得了最佳的表现。同时,如图5所示,HRToxPred在Tox21的雷达图中拥有最大的面积。
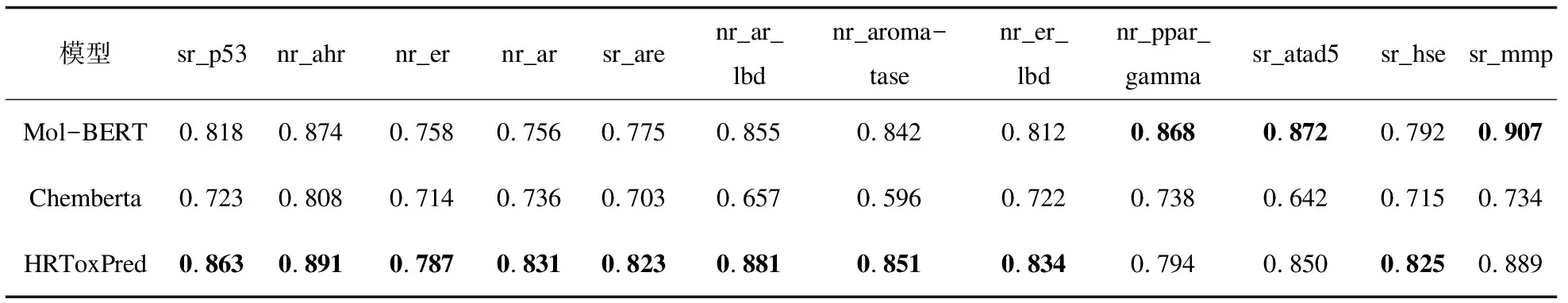
表6 对比实验结果比较(ROC-AUC↑)
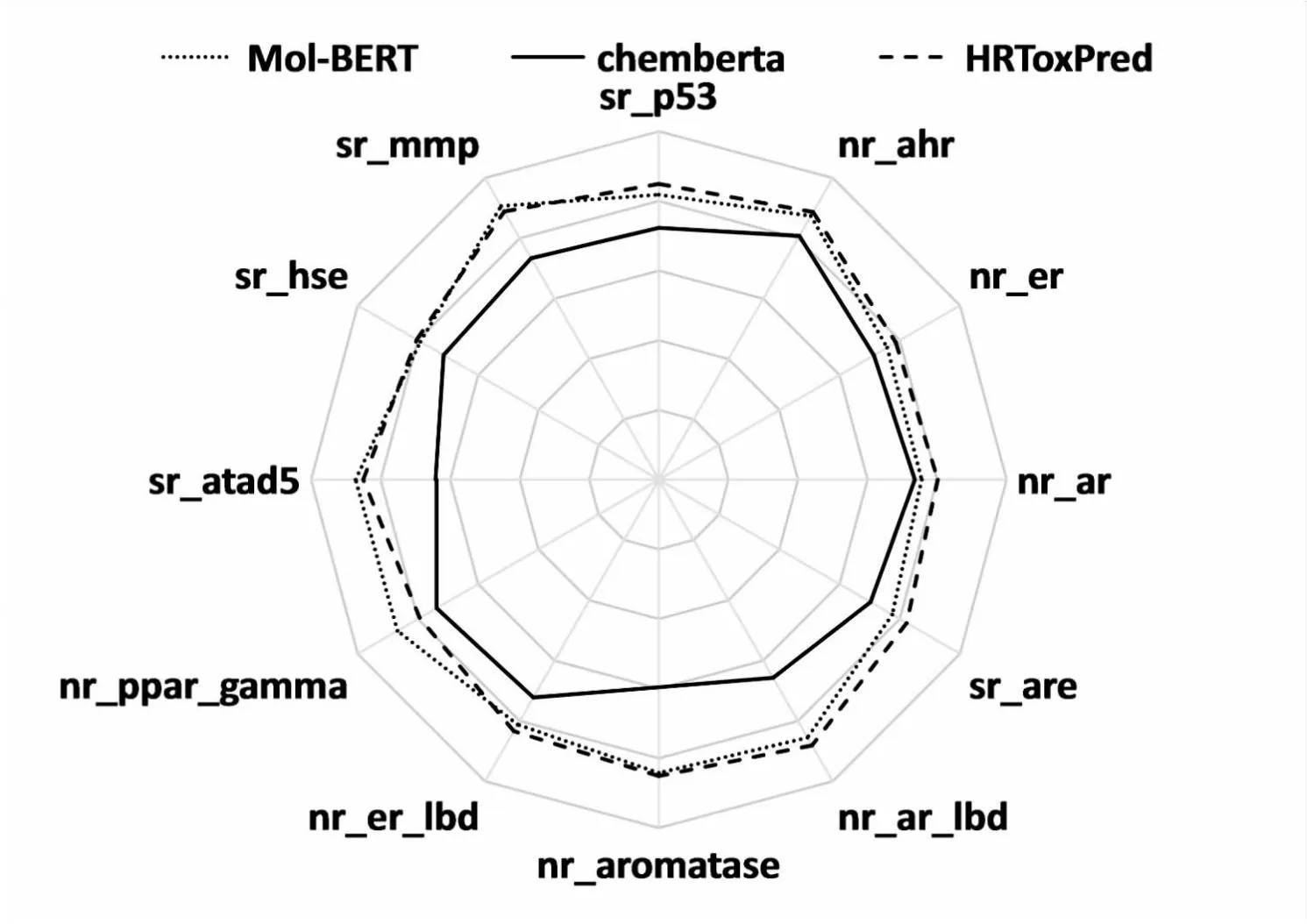
图5 Tox21各任务AUC分数雷达图
4 结束语
该文提出了引入摩根指纹半径以及显式引入氢原子的知识增强方法,并预训练了一个原子知识增强的分子毒性预测模型HRToxPred。该模型与现有的基于序列的分子毒性预测模型有两个方面的优势:一方面是显式地引入了氢原子的知识,使得MFI字典规模更小,进而减少了模型参数量并提升了毒性预测的性能;另一方面是额外引入了摩根指纹半径的知识,增强了MFI序列的顺序特征。从对比实验和消融实验的结果可见,这两方面能使模型性能有了显著提升。
虽然该方法在一些毒性预测任务数据集上展现了良好的性能,但仍有一些局限性。从消融实验的结果来看,HRToxPred在Tox21数据集中nr_aromatase,nr_ppar_gamma,sr_atad5,sr_mmp等任务上的表现不如预期,后续工作会结合深度学习理论和结构化学的知识来详细分析引入氢原子及半径对不同毒性预测任务的影响。此外,根据化学的领域知识可知,分子中的官能团对分子的特性有较大影响[20],因此后续工作会考虑引入相关领域知识。
