姜黄素-生物素偶联物的合成及抗肿瘤活性研究
2024-03-23马红艳王立强
马红艳,王立强
(1.深圳市龙华区中心医院 药学部,广东 深圳 518110; 2.华侨大学 生物医学学院,福建 泉州 362021)
姜黄素(curcumin)是具有β-二酮的庚二烯与两个邻甲基化的酚相连组成的对称分子结构的多酚类化合物,为天然植物姜黄(Curcuma Longa L.)、莪术(Curcuma zedoaria(Christm.)Rosc.)、郁金(Curcuma aromatica Salisb.)等姜黄属植物中的有效成分之一。具有抗炎、抗氧化、抗肿瘤等广泛药理活性作用[1-4]。研究表明,姜黄素在体内有一定耐受性,对胃肠道刺激小,且毒性较低,对肿瘤细胞增殖有一定抑制作用,因此被国内外学者作为抗肿瘤天然药物的候选药物[5-9]。但由于姜黄素存在:生物体内环境中溶解性差[10-11]、体内代谢快和生物利用度低[12]等不足,在临床应用中受到一定限制。因此通过对姜黄素进行结构修饰来提高其生物活性、改善其溶解性,为姜黄素类新药研发提供参考依据。
生物素(Biotin)又称维生素H、辅酶R,在自然界来源广泛且较容易制备,属于水溶性的B族维生素。肿瘤细胞表面的生物素受体含量明显高于其他正常组织,开发具有生物素及其衍生物的配体可以选择亲和该受体部位,减少抗肿瘤药物在非靶部位的蓄积和滞留,使药物具有靶向特异性[13-15]。本文考虑将姜黄素酚羟基进行结构修饰改造,制成一系列中间体,并将具有一定肿瘤细胞靶向特征的生物素通过酯键或酰胺键偶联,制备合成一系列姜黄素-生物素偶联物。最后通过MTT法分别评价目标化合物(2、4和5)对KB细胞增殖的抑制作用,以期为相关领域进一步研究提供参考依据。
1 实验部分
1.1 仪器与试剂
DF-101S型集热式电磁搅拌器(巩义市予华仪器有限公司);R202型旋转蒸发仪(上海申胜生物技术有限公司);CCL-170B-8型二氧化碳培养箱(新加坡艺思高科技有限公司);AC2-4S1型生物安全柜(新加坡艺思高科技有限公司);TS100-F型倒置显微镜(尼康株式会社)。
姜黄素(国药集团化学试剂有限公司);其余试剂均为分析纯;KB:人口腔表皮样癌细胞(中国科学院上海细胞库);澳洲特级胎牛血清(以色列BioInd公司);RPMI1640(Gibco公司)。
1.2 中间体化合物的合成
(1) 化合物6的合成
如图1所示,将0.30 g(0.81 mmol)姜黄素溶于45 mL丙酮溶液中,依次向溶液中加入0.23 g(1.66 mmol)碳酸钾和0.15 g(0.77 mmol)溴乙酸叔丁酯,反应加热回流8 h,反应结束后,冷却过滤,滤液减压浓缩,残余物经硅胶柱色谱分离纯化[16],得0.15 g黄色固体化合物6,产率36.90%; m.p.86.2~89.4 ℃,分子式C27H30O8; HR-MS(ESI)(CH3CN,ES-API):m/z483.3{[M+H+]};1H NMR(400 MHz,CDCl3),δ:7.60(d,J=15.7 Hz,2H),7.16~7.08(m,3H),7.06(s,1H),6.94(d,J=8.3 Hz,1H),6.77(d,J=8.5 Hz,1H),6.51(d,J=5.7 Hz,1H),6.47(d,J=5.9 Hz,1H),5.84(d,J=18.4 Hz,2H),4.63(s,2H),3.95(d,J=5.1 Hz,6H),1.48(s,9H);13C NMR(100MHz,CDCl3),δ:183.7,182.9,167.558,149.7,149.3,148.0,146.8,140.7,140.1,129.2,127.7,122.9,122.5,122.0,121.8,114.9,113.3,110.8,109.7,101.3,82.6,66.3,56.0,55.9,28.1; IR(KBr),ν:3412,3060,2980,2930,1750,1627,1586,1464,1425,1509,1140,1032,968,848,817 cm-1。
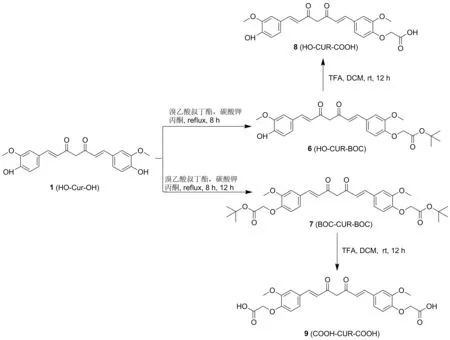
图1 中间体化合物6、7、8和 9的合成路线
(2) 化合物7的合成
如图1所示,将1.0g(2.72 mmol)姜黄素溶于80 mL丙酮中,依次向溶液中加入3.0 g(21.71 mmol)碳酸钾和1.05 g(5.38 mmol)溴乙酸叔丁酯,加热回流反应12 h,反应结束后,冷却过滤,滤液减压浓缩,残余物经硅胶柱色谱分离纯化[16],得0.70 g黄色固体化合物7,产率43.36%; m.p.120.8~124.5 ℃,分子式C33H40O10; HR-MS(ESI)(CH3CN,APCI):m/z597.3{[M+H+]};1H NMR(400 MHz,CDCl3),δ:7.60(d,J=15.7 Hz,2H),7.11(d,J=7.0 Hz,4H),6.77(d,J=8.7 Hz,2H),6.51(d,J=15.8 Hz,2H),5.82(s,1H),4.63(s,4H),3.94(s,6H),1.48(s,18H);13C NMR(100MHz,CDCl3),δ:183.2,167.6,149.2,149.1,140.2,128.3,122.4,113.1,111.2,101.4,81.5,65.4,55.7,27.7; IR(KBr),ν:2980,2936,1754,1630,1586,1464,1420,1509,1032,968,846,805 cm-1。
(3) 化合物8的合成
如图1所示,将0.14g(0.29 mmol) 化合物6溶于10 mL DCM中,加入1 mL(1.54g,13.51 mmol)三氟乙酸,室温反应12 h,减压浓缩得粗品,粗品经硅胶柱色谱分离纯化[17]得0.08g黄色固体化合物8,产率64.83%; m.p.90.3~93.5℃,分子式C23H22O8;HR-MS(ESI)(CH3CN,APCI)m/z:428.2{[M+H+]};1H NMR(400 MHz,DMSO-d6),δ:13.05(s,1H),9.68(s,1H),7.57(d,J=15.8 Hz,2H),7.34(t,J=14.8 Hz,2H),7.23(d,J=8.6 Hz,1H),7.16(d,J=8.0 Hz,1H),6.92(t,J=13.4 Hz,1H),6.86~6.81(m,2H),6.77(d,J=15.8 Hz,1H),6.09(s,1H),5.76(s,1H),4.73(d,J=14.0 Hz,2H),3.83(t,J=8.6 Hz,6H);13C NMR(100 MHz,DMSO-d6),δ:183.8,182.5,167.5,149.4,149.1,148.0,141.0,139.9,128.4,126.3,123.2,122.4,121.1,115.7,113.1,111.4,111.2,101.0,81.5,65.4,55.7,55.6,27.7; IR(KBr),ν:3396,2946,1742 1627,1586,1425,1509,1029,968,848,817 cm-1。
(4) 化合物9的合成
如图1所示,将0.70g(1.18 mmol)化合物7溶于35 mL DCM中,加入7 mL(1.54 g,94.56 mmol) 三氟乙酸,室温反应约12 h,减压浓缩,经硅胶柱色谱分离纯化[17],得0.08 g黄色固体化合物9。产率78.10%; m.p.201.3~203.7℃,分子式C25H24O10; HR-MS(ESI)(CH3OH,ES-API)m/z:483.2{[M-H]+};1H NMR(400 MHz,DMSO-d6,δ:13.06(s,2H),7.95(s,1H),7.59(d,J=15.8 Hz,2H),7.38(s,2H),7.24(d,J=8.4 Hz,2H),6.88(dd,J=21.4,12.1 Hz,4H),6.12(s,1H),4.75(s,4H),3.85(s,6H);13C NMR(100MHz,DMSO-d6),δ:183.2,169.9,149.3,149.1,140.3,128.2,122.5,122.4,112.9,111.1,101.0,64.9,55.7; IR(KBr),ν:3396,2946,1742,1627,1586,1425,1509,1029,968,848,817 cm-1。
(5) 化合物10的合成
如图2所示,冰浴下,将40 mL含Boc2O的THF溶液缓慢加到60 mL含有4.44 g 1,8-二氨基-3,6-二氧杂辛烷和1.40 mL三乙胺的THF溶液中,室温反应24 h。反应液减压浓缩加入40 mL水,并用75 mL正己烷洗水层,经100 mL DCM萃取后,有机层减压浓缩,经过硅胶柱色谱分离纯化,得2.02 g无色液体化合物10,产率80.70%;分子式C11H24N2O4; IR(KBr),ν:3355,2982,2936,2874,1696,1520,1177,1131 cm-1。
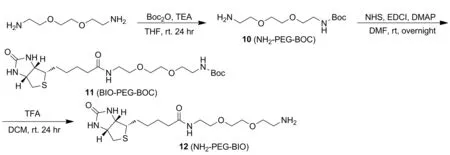
图2 中间体化合物10、11和12及目标化合物4和5的合成路线
(6) 化合物11的合成
(7) 化合物12的合成
如图2所示,将0.50 mL三氟乙酸加入到2.50 mL含0.10 g化合物11的DCM溶液中,室温反应24 h,减压浓缩至干的粗品,粗品经过硅胶柱色谱分离纯化,得到0.07 g化合物12,无色油状液体,产率81.33%;分子式C16H30N4O4S,1H NMR(400 MHz,DMSO-d6),δ:7.86(t,J=5.5 Hz,1H),6.42(s,1H),6.38(s,1H),4.37~4.26(m,1H),4.13(s,1H),3.63~3.48(m,5H),3.40(t,J=6.0 Hz,4H),3.19(q,J=5.8 Hz,2H),3.09(t,J=9.3 Hz,1H),2.97(t,J=5.2 Hz,2H),2.82(dd,J=12.4 Hz,5.0 Hz,1H),2.58(d,J=12.3 Hz,1H),2.07(t,J=7.4 Hz,2H),1.50(ddd,J=27.9 Hz,20.2 Hz,7.7 Hz,4H),1.29(dd,J=20.9 Hz,14.5 Hz,2H);13C NMR(101 MHz,DMSO-d6),δ:172.2,162.7,69.6,69.4,69.1,66.8,61.0,59.2,55.4,38.7,38.3,35.1,28.1,25.2.IR(KBr),ν:3366,2982,2936,2864,1698,1520,1456,1392,1177 cm-1。
1.3 目标化合物的合成
(1) 化合物2的合成
如图3所示,将0.36 g(1.48 mmol)生物素溶于10 mL DMF中,室温搅拌至全溶,依次加入0.39 g(1.89 mmol)DCC、0.30g(2.46 mmol)DMAP和0.63 g(1.63 mmol)姜黄素,室温反应24~36 h。反应结束后,过滤,溶液减压浓缩后加入适量二氯甲烷,有机层依次用饱和NH4Cl、水、饱和食盐水洗涤,有机层经无水硫酸钠干燥,浓缩得粗品,粗品经硅胶柱色谱分离纯化,得0.39 g化合物2:黄色固体,产率44.37%; m.p.182~183 ℃,分子式C31H34N2O8S; HR-MS(ESI)(CH3CN,ES-API)m/z:298.2{[(M+2H)/2]+};1H NMR(400 MHz,DMSO-d6),δ:9.71(s,1H),7.60(dd,J=15.8,3.6 Hz,2H),7.50(s,1H),7.37~7.27(m,2H),7.16(t,J=9.9 Hz,2H),6.96(d,J=16.0 Hz,1H),6.81(t,J=12.8 Hz,2H),6.47(s,1H),6.38(s,1H),6.13(s,1H),4.31(d,J=5.2 Hz,1H),4.17(s,1H),3.84(s,6H),3.15(s,1H),2.85(dd,J=12.4 Hz,4.7 Hz,1H),2.61(d,J=4.3 Hz,1H),2.58(s,2H),1.74~1.30(m,6H);13C NMR(101 MHz,DMSO-d6),δ:184.9,181.4,171.0,162.7,151.2,149.5,148.0,141.5,140.8,139.0,133.8,126.2,124.5,123.3,121.2,115.7,111.9,111.4,101.3,61.0,59.2,56.0,55.7,55.4,33.0,28.0,24.5; IR(KBr),ν/cm-1:3361,3304,2931,2865,1760,1690,1630,1640,1586,1509,1029,973。
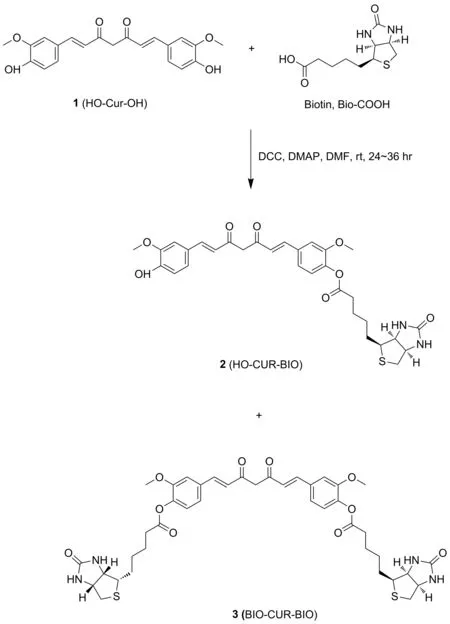
图3 目标化合物2的合成路线
(2) 化合物4的合成
如图2所示,向18 mL 含0.08 g(0.21 mmol)化合物12和0.08 g(0.19 mmol)化合物8的DMF溶液中依次加入0.05 g(0.26 mmol)DCC,0.03 g(0.26 mmol)NHS和0.03 g(0.26 mmol)DMAP,室温反应24 h。反应结束后,减压浓缩,加入40 mL二氯甲烷,依次用HCl(0.1M)、饱和食盐水洗涤有机层,干燥浓缩得粗品,粗品经硅胶柱色谱分离纯化,得0.12 g化合物4:黄色固体,产率81.56%; m.p.95.2~98.3℃,分子式C39H50N4O11S,HR-MS(ESI)(CH3OH,ES-API):m/z392.2{[(M+2H)/2]+},1H NMR(400 MHz,DMSO-d6),δ:9.68(s,1H),7.95(t,J=5.5 Hz,1H),7.82(s,1H),7.56(d,J=15.8 Hz,2H),7.38(s,1H),7.32(s,1H),7.28~7.18(m,1H),7.17~7.08(m,1H),6.94(d,J=8.3 Hz,1H),6.83(ddd,J=33.4 Hz,18.1 Hz,9.7 Hz,3H),6.41(s,1H),6.35(s,1H),6.08(s,1H),4.52(d,J=13.6 Hz,2H),4.31~4.24(m,1H),4.10(s,2H),3.82(dd,J=15.1 Hz,9.8 Hz,6H),3.53~3.40(m,8H),3.21~3.11(m,4H),3.06(t,J=9.4 Hz,1H),2.79(dd,J=12.4 Hz,5.1 Hz,1H),2.56(d,J=12.4 Hz,1H),2.05(t,J=7.3 Hz,2H),1.59~1.25(m,6H);13C NMR(100 MHz,DMSO-d6),δ:184.0,182.4,172.1,167.5,162.7,149.4,149.3,149.2,148.0,141.1,139.8,128.7,126.3,123.2,122.6,122.4,121.1,115.7,114.0,111.4,111.0,101.0,69.5,69.2,68.8,68.0,63.1,61.0,59.2,55.7,55.6,55.4,48.6,38.4,38.3,35.1,28.2,28.0,25.2; IR(KBr),ν:3290,2931,2862,1690,1627,1586,1509,1272,1137,1029,968,848,815 cm-1。
(3) 化合物5的合成
如图2所示,向18 mL含0.11 g(0.30 mmol)化合物12的DMF溶液中依次加入0.07 g(0.14 mmol)化合物9,0.07 g(0.35 mmol)DCC,0.04g(0.35 mmol)NHS,0.04 g(0.35 mmol)DMAP,室温反应24 h。反应结束后,减压浓缩,加入40 mL二氯甲烷,依次用HCL(0.1 M)、饱和食盐水洗涤有机层,有机层浓缩得粗产品,粗品经硅胶柱色谱分离纯化,得0.08 g化合物5:黄色固体,产率44.56%; m.p.135.3~136.1 ℃,分子式C57H80N8O16S2,HR-MS(CH3OH,ES-API)m/z:599.3 {[(M+2H)/2]+};1H NMR(400 MHz,DMSO-d6),δ:7.98(s,2H),7.85(s,2H),7.59(d,J=15.9 Hz,2H),7.45~7.19(m,4H),6.99~6.73(m,4H),6.42(s,2H),6.36(s,2H),6.13(s,1H),4.54(d,J=11.1 Hz,4H),4.34~4.25(m,2H),4.12(s,2H),3.87(s,6H),3.56~3.45(m,12H),3.39(d,J=6.0 Hz,8H),3.18(dd,J=11.3 Hz,5.6 Hz,4H),3.08(t,J=9.3 Hz,2H),2.81(dd,J=12.4 Hz,4.9 Hz,2H),2.57(d,J=12.4 Hz,2H),2.06(t,J=7.3 Hz,4H),1.63~1.42(m,8H),1.26(dd,J=15.4 Hz,7.8 Hz,4H);13C NMR(100 MHz,DMSO-d6),δ:183.2,172.1,167.5,162.7,149.3,149.4,140.2,128.7,122.6,122.5,114.0,111.1,101.1,69.5,69.2,68.8,68.0,61.0,59.2,55.7,55.4,38.4,38.3,35.1,28.1,28.0,25.2; IR(KBr),ν/cm-1:3290,3084,2926,2870,1670,1553,1509,1272,1137,1029,968,848,815。
1.4 生物素类姜黄素衍生物的初步药理活性分析
(1) 细胞培养
KB细胞的培养条件为含体积分数为10%的胎牛血清RPMI 1640、青霉素(100 IU·mL-1)和链霉素(100 μg·mL-1),在37 ℃,5% CO2孵育箱中培养。待细胞密度长到70%~90%,取对数生长期细胞用于抑制率测定实验。
(2) MTT法评价姜黄素衍生物对KB细胞增殖的抑制作用
在96孔板上,接种KB细胞,接种量5000个/孔,每孔200 μL;待细胞贴壁后,实验组分别加入不同浓度的姜黄素衍生物(100 μmol/L,50 μmol/L,25 μmol/L,12.50 μmol/L,6.25 μmol/L),对照组加入含相同体积DMSO的新鲜培养基,每组设5个复孔,37 ℃培养24 h;去除含药培养基,每孔用PBS洗涤2次,加入180 μL新鲜培养基,避光加入20 μL MTT; 37 ℃培养4 h后,取出并弃去上清液,加入150 μL DMSO,振荡10 min,酶标仪570 nm检测OD值,通过下面的公式,得到不同浓度下的抑制率;以浓度为横坐标,以抑制率为纵坐标,绘制生长抑制曲线,求出该药物对细胞生长抑制率为50%的浓度即IC50值。
细胞抑制率%=(对照组OD值-加药组OD值)/对照组OD值×100%。
2 结果与讨论
2.1 目标化合物的化学合成
(1) 通过连接水溶性连接臂得到的姜黄素杂合物
该方法对具有较好水溶性的商品化1,8-二氨基-3,6-二氧杂辛烷进行单边Boc保护,得到化合物10,再与带有羧基的生物素在碳二亚胺类化合物(DCC或EDCI)和DMAP催化下以较高的产率得到化合物11。化合物11在含有20%TFA的二氯甲烷溶液中脱除N-Boc保护基,获得含氨基官能团的化合物12,与经NHS活化的化合物8和9在DCC、DMAP条件下缩合,最后得到了生物素标记的姜黄素衍生物4和5。
(2) 姜黄素衍生物合成过程副反应的控制
姜黄素中2个酚羟基和一个亚甲基在碱性条件下都可以和卤代烷反应,但由于酚羟基比酮的α-位碳的反应活性更强,采用二碳酸二叔丁酯将酚羟基保护成碳酸叔丁酯,产率高,产品纯度好,而且脱保护的产率也较高。另外,采用NHS/DMAP/DCC活化姜黄素羧基时间过长,可能会引起羧基化合物的自身缩合等副反应的发生,因此控制羧基活化时间成为反应的关键。而反应物溶剂的选择是反应能否顺利进行和提高产率的重要因素,采用DMF溶剂可以使反应物持续向目标反应物顺利进行,从而提高产率。
2.2 抗肿瘤活性分析
采用MTT法初步研究了合成的相关目标化合物(2、4、5)的抗肿瘤活性,结果见表1。随着化合物2作用时间的延长,其对KB细胞的IC50值逐渐减少,初步表明其细胞毒作用具有一定时间依赖性且抗肿瘤活性优于原药姜黄素。化合物4作用时间为24~48 h,IC50值逐渐增大,表明其抑制作用随着时间逐渐减弱,其作用效果大致与原药姜黄素相同。而双生物素修饰的姜黄素衍生物BIO-PB-CUR-PB-BIO则在100 μmol/L浓度内没有观察到抗肿瘤活性。
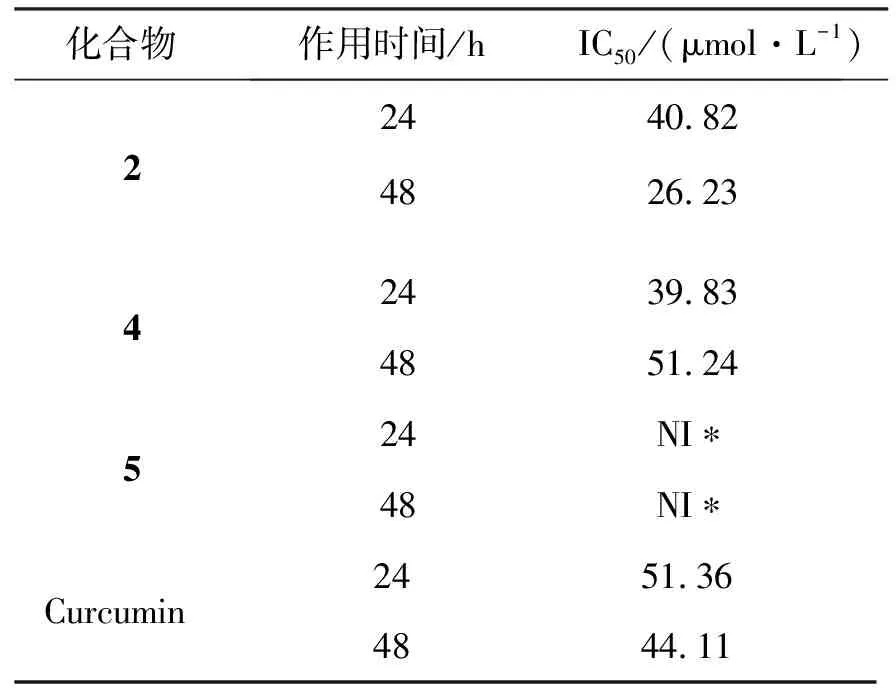
表1 目标化合物不同时间下的IC50值
3 结论
本研究制备得到生物素标记姜黄素衍生物。首先将生物素羧基在氯化亚砜作用下制备成酰氯,再与姜黄素酚羟基进行酯化反应,得化合物2。另外以姜黄素2端酚羟基为修饰位点,合成姜黄素羧基化衍生物;同时合成了具有游离氨基的寡聚乙二醇-生物素中间体,将其与将姜黄素羧基化衍生物DCCDMAP条件下进行酰胺化反应,得到化合物4和5。通过MTT法初对所制备化合物进行初步细胞活性考察,显示化合物2在作用24 h及48 h时对KB细胞的IC50分别是40.82 μmol/L和26.23 μmol/L,提示其抑优于抗肿瘤活性药姜黄素。化合物4在作用24 h及48 h时对KB细胞的IC50分别是39.83 μmol/L和51.24 μmol/L,初步认为将姜黄素与生物素进行偶联生成酯化与酰胺化产物具有较姜黄素本身更强的抗肿瘤细胞活性,为姜黄素类新药在抗肿瘤临床应用开发提供依据。
