坏死性凋亡相关的长链非编码RNAs生物标志物预测肝细胞癌预后模型的构建
2024-03-20黄琼庆梁珍贵黄琪琪欧超
黄琼庆 梁珍贵 黄琪琪 欧超
作者单位:530000 南宁 广西医科大学附属肿瘤医院检验科
肝细胞癌(hepatocellular carcinoma,HCC)是原发性肝癌的主要类型[1],主要采用以手术为主并结合靶向治疗、介入放射治疗、中医药治疗等的综合疗法[2]。随着靶向治疗和免疫治疗在HCC 研究中不断突破,其治疗也取得了较大进展[3]。但HCC 患者的整体长期生存率仍然不容乐观,5 年生存率仅约为18%[4]。而且,HCC 高复发率一直是临床研究的难题。因此,寻找更有效的肝癌预测标志物对监测其复发和揭示新的治疗靶点至关重要。
坏死性凋亡是一种以Caspase 非依赖的方式调节的坏死性细胞死亡,主要由受体相互作用蛋白激酶1(receptor-interacting protein kinase 1,RIPK1)、受体相互作用蛋白激酶3(RIPK3)和混交激酶域蛋白(mixed lineage kinase domain-like protein,MLKL)介导,并被坏死性凋亡抑制剂(necrostatin-1,NEC-1)抑制,在肿瘤发生、肿瘤转移、抗肿瘤免疫等肿瘤生物学调节中发挥重要作用[5-6]。人类基因组只有大约2%的RNA被编码成蛋白质。在非编码RNAs 中,超过200 个碱基的被定义为长链非编码RNAs(long non-coding RNAs,lncRNAs)。在HCC 中,lncRNAs 可以通过招募、引导等在表观遗传、转录和转录后水平调节基因的表达,并参与肿瘤的增殖、凋亡、转移和其他生物学活动[7-8]。因此,进一步阐明坏死性凋亡相关的长链非编码RNAs(necroptosis-related long non-coding RNAs,NRLs)与HCC的关系对探索HCC的新治疗靶点和改善患者预后具有重要意义。然而,目前NRLs在HCC中的研究有限。
本研究基于NRLs 构建HCC 风险预后模型,并进一步探索可能的作用机制。在治疗方面,本研究分析了不同风险组之间免疫浸润和免疫检查点的差异,以期为HCC患者应用免疫治疗提供理论基础。
1 资料与方法
1.1 数据集
从TCGA 数据库(https://portal.gdc.cancer.gov)获得转录数据RNA 序列和临床数据(TCGA-LIHC)。随后,在GSEA(http://www.gsea-msigdb.org/gsea/index.jsp)获得坏死性凋亡相关基因(necrosis-related genes,NRG)集,并结合以往文献[9],共获得67 个NRG。随后通过Pearson 相关分析67 个NRG 与所有lncRNAs的表达水平之间的相关性,获得NRLs(r>0.4 和P<0.001)。筛选条件:|log2FC|>1 和P<0.05。采用“LIMMA”包和“BiocManager ”包分析NRLs 在HCC 癌组织及癌旁组织中的表达,并绘制火山图。
1.2 NRLs风险模型的构建
获得343 例具有完整生存信息的HCC 样本,将整个数据集按1∶1 的比例分为训练集和测试集,其中训练集用于构建风险模型,测试集用于验证风险模型。通过单因素Cox 回归分析获得与HCC 患者预后相关的NRLs,然后通过最小绝对收缩和选择算法(least absolute shrinkage and selection operator,LASSO)和多因素Cox回归构建风险评分预后模型。
1.3 NRLs风险模型的评估
采用“timeROC”包绘制受试者工作特征(receiver operating characteristic,ROC)曲线,并计算ROC 曲线下面积(area under curve,AUC)评估模型的区分度。采用“survival”和“survminer”包进行生存分析。根据训练集中的风险评分中位数将患者分为高风险组、低风险组,并采用Log-rank 检验比较高风险组、低风险组之间总生存期(overall survival,OS)的差异。采用多因素Cox 回归分析,确定风险评分是否可作为预测肝细胞癌患者预后的潜在独立指标。
1.4 生信分析
采用GSEA(Kegg.v7.5.1,symbs.GMT)进行通路富集分析,以识别高风险人群中显著活跃的信号通路(P<0.05 和FDR<0.05)。肿瘤微环境分析方面,使用单一样本GSEA 对HCC 中的免疫细胞和免疫功能评分,并用“GSVA”包对其进行定量分析。最后使用“ggpubr”包比较不同亚型患者的免疫检查点激活情况。
1.5 qRT-PCR检测NRLs在HCC细胞中的表达水平
用Trizol 试剂(Invitgen,美国)提取纯化的细胞总RNA。使用RR047A(Takara,日本)试剂进行逆转录。使用荧光定量聚合酶链式反应试剂盒(ChamQUniversal SYBR QPCR Master Mix)(Vazyme,中国)和PCR扩增仪(ABI7500,美国)进行荧光定量聚合酶链式反应。反应体系:2'ChamQ Universal SYBR qPCRMaster Mix 10 mL、Primer1(10 mmol/L)0.4 mL、Primer2(10 mmol/L)0.4 mL、Template DNA 2 mL、ddH2O 7.2 mL。PCR扩增仪设置实验条件:预变性95 ℃30 s、变性95 ℃5s、退火60 ℃30 s,40 个循环。以β-actin为内参,采用2-ΔΔCT法计算各细胞中NRLs 的表达量。引物序列如下:AL117336.2forward primer 5′- TGTTGCTCTGGCTGATCTTGAACTC-3′ ,reverse primer 5′-GCTAGGTGTGGTGGCTCATACTTG-3′;LINC01224forwardprimer 5′-CCAGGTCTTCAGATTCCACCACTTG-3′,reverse primer 5′-AGAAAGGCGGGACAACTCAAAGC-3′ ;FOXD2-AS1forward primer 5′- TGGGTTGAGGGTCTGTGACTGTAG-3′,reverse primer 5′-GCTGCCGCTGGAGTATTCTTGG -3′;MKLN1-ASforward primer 5′-TGGTGGTGTTTCTCTCTGAAAGCAG-3′,reverse primer 5′- ATGGCAGCGGAGTCCTCAAGG -3′;β-actinforward primer 5′-GACCTGACTGACTACCTCATGAAGAT -3′,reverse primer 5′-GTCACACTTCATGATGGAGTTGAAGG-3′。
1.6 统计学方法
数据的处理与分析由R 4.2.1 软件通过合适的软件包完成。计量资料的多组间比较采用单因素方差分析,两组比较采用独立样本t检验。以双侧P<0.05为差异有统计学意义。
2 结果
2.1 差异表达的NRLs
从TCGA 数据库中获得424 例HCC 患者的转录RNA 序列和临床病理信息,其中343 例具有完整临床特征的样本用于后续分析。对67个NRG 进行共表达分析,最终得到了791 个NRLs(r>0.4 和P<0.001),见图1A。火山图显示,791个NRLs中有9个表达下调,其余表达均上调(均P<0.001),见图1B。
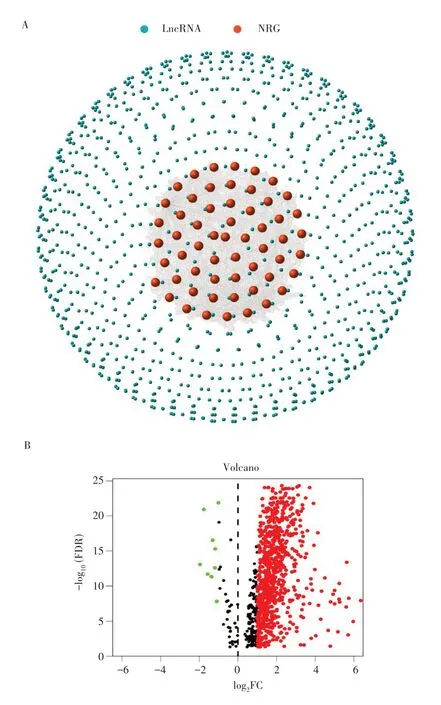
图1 NRLs的差异表达Fig.1 Differential expression of NRLs
2.2 NRLs风险模型的构建
单因素Cox回归筛选出17个与HCC患者OS相关的NRLs(均P<0.001),见图2A。17个NRLs在HCC组织中的表达水平均高于正常组织,见图2B。将343例HCC 样本按1∶1 的比例随机分为训练集(n=172)和测试集(n=171),训练集和测试集的一般资料详见表1。随后在训练集中,采用LASSO-Cox 回归分析,同时设置10倍交叉验证,以获得最优模型,见图2C、图2D。该模型由4个NRLs(MKLN1-AS、FOXD2-AS1、AL117336.2、LINC01224)组成。HCC 患者的风险评分=1.758×MKLN1-AS+0.322×FOXD2-AS1+0.698×AL117336.2+0.585×LINC01224。

表1 训练集和测试集的一般资料Tab.1 General information on training and testing sets
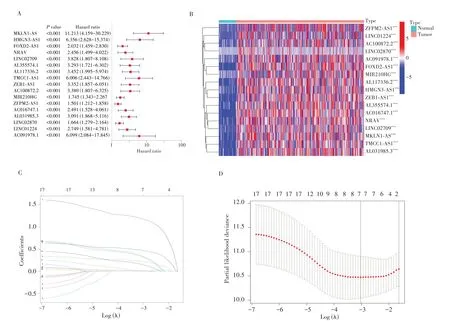
图2 NRLs预测模型的构建Fig.2 Construction of predictive model base on NRLs
2.3 NRLs风险模型的评估
基于训练集的多因素Cox 回归分析显示,校正其他临床病理特征后,风险评分是独立预后因素(HR=1.275,P<0.001),见图3A。在训练集中,风险模型预测1、3、5 年OS 的AUC 分别为0.816、0.747、0.752,在验证集中分别为0.713、0.621、0.626,见图3B、图3C。根据风险评分中位数(0.865)将训练集和测试集的患者分为高风险组、低风险组。散点图显示,高风险区域的红点更密集,见图4A。提示高风险组患者具有更高的风险评分、更短的生存时间。生存分析显示,在训练集和测试集中,高风险组的OS 均低于低风险组(均P<0.05),见图4B。
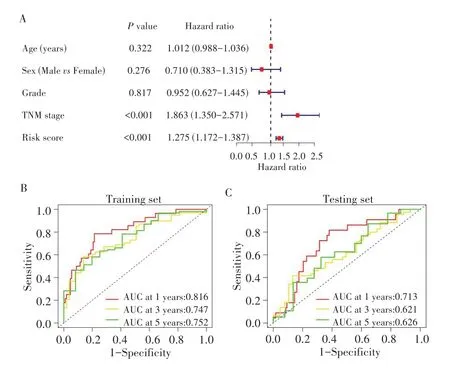
图3 NRLs风险模型的预测价值Fig.3 Predictive value of NRLs risk models

图4 不同风险组患者的预后差异Fig.4 Prognostic differences among different risk groups
2.4 通路富集与免疫相关功能的差异分析
利用GSEA 软件筛选出10 条在高风险人群中功能显著丰富的信号通路(均P<0.05,FDR<0.05)。其中与免疫功能相关的有3 条信号通路:包括内吞作用、囊泡转运中的相互作用和黏附连接,其余7 条信号通路均为癌症相关信号通路,见图5A。此外,使用ssGSEA 量化免疫细胞、免疫功能与风险模型之间的关系。结果发现高风险组的巨噬细胞、T 调节细胞(Treg)和MHC-Ⅰ类分子的表达水平更高,免疫得分更高;而低风险组的B 细胞、中性粒细胞、NK 细胞、肥大细胞、Ⅰ型和Ⅱ型干扰素应答表达水平更高,免疫得分更高,见图5B、图5C。所有免疫检查点在高风险组中都显示出较高的活性,如TNFRSF14、CD27 和LGALS9(图5D),这意味着高风险组患者的免疫活性更高,对免疫治疗更敏感。提示可根据HCC 患者的NRLs 风险模型分组选择合适的免疫检查点抑制剂来治疗。
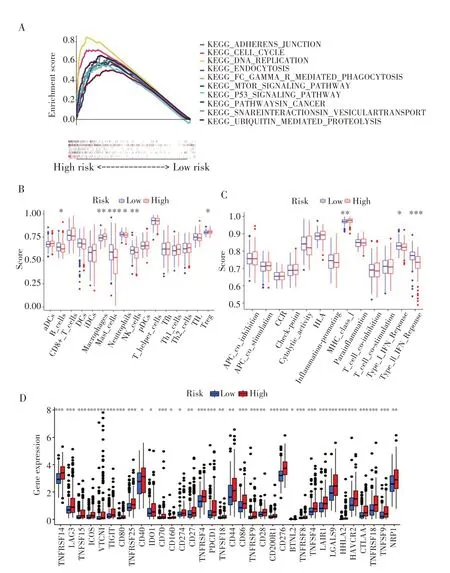
图5 通路富集和免疫相关功能分析Fig.5 Pathway enrichment and immune-related function analysis
2.5 NRLs在HCC细胞中呈高表达
qRT-PCR 结果(图6)显示,在正常肝细胞MIHA和HCC细胞Huh7中检测NRLs的表达,结果显示,4 个NRLs(MKLN1-AS、FOXD2-AS1、AL117336.2、LINC01224)在HCC细胞中均表达上调(均P<0.01)。

图6 NRLs在MIHA和Huh7细胞中的表达差异Fig.6 Differential expression of NRLs in MIHA and Huh7 cells
3 讨论
目前,HCC 患者的OS 往往因为复发、转移而缩短[10]。因此,提前评估患者的预后信息对选择适合的治疗方案和随访方案具有重要价值。最近的研究发现,坏死性凋亡与HCC进展相关,例如在HCC 细胞中引入过量的山梨糖醇脱氢酶,发现其可通过增强坏死性凋亡信号来抑制肿瘤生长和干性[11]。因此,坏死性凋亡相关途径可能是肿瘤治疗的潜在靶点,但目前NRLs 在HCC 中的作用机制尚不清楚,因此构建可靠的HCC预后预测模型可能成为改善预后的关键。
本研究筛选出对HCC预后有价值的4个NRLs(包括MKLN1-AS、FOXD2-AS1、AL117336.2 和LINC01224)来构建NRLs 风险模型。根据现有文献,MKLN1-AS在HCC 发展中起促进作用[12]。而FOXD2-AS1 可通过调节miR-206/MAP3K1 轴而加速HCC 进展[13]。LINC01224 则通过与miR-485-5p 特异性结合促进咽鳞状细胞细胞癌的增殖和侵袭能力[14],但LINC01224在HCC中的作用机制尚未被报道。AL117336.2在既往研究中未见报道,该基因可能是HCC潜在的治疗靶点。
为证明NRLs 风险模型的实用价值,本研究进一步将所有的HCC 样本以1∶1 比例随机分配到训练集和测试集中,并根据中位风险评分将患者分为高风险组、低风险组,以验证所构建的NRLs风险模型的预后价值。本研究结果显示低风险组患者的预后优于高风险组,此外在校正潜在混杂因素后,多因素Cox回归表明该风险评分是预测HCC 患者生存的独立预后因素,ROC 曲线表明本研究构建的NRLs 风险模型对患者的1、3、5 年OS 具有较好的预测能力。
此外,本研究还筛选出10 条在高危人群中功能显著丰富的通路,这为后续的机制研究提供了方向。越来越多的证据表明,肿瘤细胞的坏死性凋亡通过在肿瘤微环境中释放细胞因子和趋化因子来驱动癌细胞和免疫细胞之间的相互作用,从而参与癌症相关的免疫反应[15-16]。本研究发现不同风险组人群的免疫状态不同,其中低风险组的免疫细胞(即B 细胞、中性粒细胞、NK 细胞、肥大细胞)和免疫功能(即Ⅰ型和Ⅱ型干扰素应答)处于活跃状态,在高风险人群中,免疫细胞(即巨噬细胞、T 调节细胞)和免疫功能(即MHC-Ⅰ类)处于活跃状态。先前的研究表明,T 细胞和NK 细胞在HCC 中浸润增加是一个有利的预后因素[17-19];而T 调节细胞浸润增加的免疫微环境的肿瘤往往预后较差[20-21]。
现有的报道显示,不同的免疫状态与肿瘤微环境有关,从而导致不同的预后和免疫治疗反应[22]。在HCC和其他癌症中,免疫检查点通过在肿瘤和间充质细胞中表达相应的配体来逃避抗肿瘤免疫反应。一些抗免疫检查点疗法,如程序性细胞死亡蛋白1 或细胞毒性T 淋巴细胞相关蛋白是抗肿瘤免疫所必需的免疫调节分子家族的成员,目前被批准用于治疗实体肿瘤,如黑色素瘤、肺癌、头颈癌等[23-25]。本研究根据NRLs 风险模型分析了不同分组免疫检查点的差异,提示NRLs 风险模型能为HCC 患者进行个体化免疫治疗提供参考。
本研究存在一定的局限性:⑴本研究仅在公共数据集TCGA 中获取临床样本数据,获得的数据仍有限;⑵肿瘤样本和正常样本的比例不平衡,这可能影响通路富集的分析精度;⑶本研究仅利用qRT-PCR验证4 个NLRs 在HCC 细胞中的表达,后续仍需更多的临床数据和进一步的实验进行验证。
综上,本研究构建了一个基于NRLs 的HCC 预后模型并验证了其预测能力,该模型对指导临床治疗具有潜在的价值。此外,模型中的关键基因可能是HCC潜在的生物标志物。
