利用CRISPR/Cas9 技术构建Pmepa1 基因敲除的TCMK1 小鼠肾小管上皮细胞系
2024-03-12张宏民龙雯劳筱清陈雯妍商雪梅王洪连王丽粟宏伟沈宏萍沈宏春
张宏民 龙雯 劳筱清 陈雯妍 商雪梅 王洪连 王丽 粟宏伟沈宏萍 沈宏春,
(1.西南医科大学中西医结合学院,泸州 646000;2.西南医科大学附属中医医院,泸州 646000)
数据表示,慢性肾病已影响全球近10%的人,是低、中等收入国家发病率和死亡率的主要原因之一[1]。肾纤维化是所有慢性肾脏疾病及终末期肾病的共同病理特征[2],主要表现为肾间质成纤维细胞的活化和细胞外基质的过度积累,导致正常肾小管和间质结构的破坏[3-4]。随着肾纤维化的进行性发展,慢性肾病患者的肾功能逐渐恶化最终导致肾衰竭[5]。因此,研究肾纤维化的机制对寻找积极有效的治疗策略以改善或延缓肾纤维化的进展尤为重要[6]。
前列腺跨膜蛋白雄激素诱导1(prostate transmembrane protein androgen induced 1,Pmepa1)又称TMEPA1、STAG1、ERG1.2 或N4wbp4,是一种I 型跨膜蛋白,可与HECT 型E3 泛素连接酶相互作用[7]。研究表明,Pmepa1 参与p53 介导的细胞凋亡和细胞生长抑制,并与多种实体肿瘤(包括肾癌、膀胱癌、乳腺癌、前列腺癌、肺癌和宫颈癌)的发生有关[8-9]。Pmepa1 作为多种信号通路的调节器,有助于维持细胞内稳态[10]。最近发现Pmepa1 通过负反馈回路抑制雄激素和TGF-β 信号传导[11],并且Pmepa1 在TGF-βI 型受体信号传导的控制中发挥作用[12]。
CRISPR/Cas9 是一种RNA 引导的核酸内切酶,通过核苷酸碱基配对特异性切割DNA 序列的基因编辑技术[13]。由于其高效、易用和准确性,CRISPR/Cas9 在基因编辑研究中被广泛应用,并在人类疾病治疗方面表现出巨大潜力[14]。CRISPR/Cas9 技术于2013 年首次用于哺乳动物细胞(人和小鼠细胞),此后被广泛应用于生物研究、生物技术、医学、农业等各种研究领域[15-16]。
本研究将基于CRISPR/Cas9 技术将Pmepa1 敲除载体转染至TCMK1 小鼠肾小管上皮细胞,联用流式细胞分选技术和PCR 扩增片段测序,筛选建立Pmepa1 敲除TCMK1 细胞系,并在敲除Pmepa1 的TCMK1 细胞中验证TGF-β1 刺激下细胞纤维化的改变,为研究Pmepa1 在肾纤维化的功能提供细胞模型。
1 材料与方法
1.1 材料
鼠源肾小管上皮细胞系TCMK1 细胞源于西南医科大学附属中医医院中西医结合实验室;pX333骨架质粒购于Addgene 公司;用于sgRNA 克隆及Real-Time PCR 引物的寡脱氧核苷酸链由上海生工生物有限公司合成;质粒提取试剂盒;胶回收试剂盒和细胞基因组DNA 提取试剂盒购自于北京天根生物有限公司;Bbs I 限制性内切酶、T4 DNA 连接酶、PNK 磷酸化酶、逆转录试剂盒、胎牛血清(Gibco)购自于Thermo Scientific 公司;Real-Time PCR 所用SYBR 预混扩增试剂购自于天津诺唯赞生物有限公司;RNA 提取试剂Trizol 购自于生工生物工程(上海);DMEM 高糖培养基购自于上海培源生物科技有限公司;Pmepa1 抗体(武汉三鹰生物技术有限公司)、GAPDH 抗体(Thermo Fisher Scientific,MA5-15738),SMAD2 抗体、SMAD3 抗体、P-SMAD2 抗体、P-SMAD3 抗体(Cell Signaling TECHNOLOGY)。
1.2 方法
1.2.1 sgRNA 设计与寡核苷酸链的合成 根据CRISPR/Cas9 工作原理,在CRISPR 在线设计网站输入鼠源Pmepa1 编码基因序列(https://zlab.bio/guide-design-resources),搜寻符合“G(N)19NGG”模式的sgRNA 靶点序列(其中NGG 为PAM 序列),并保证所选sgRNA 序列符合以下要求:1)所选靶点位于外显子区段;2)靶点所选外显子为所有已发现Pmepa1 选择性间接体所共有。最终选择靶点位于第2 号外显子,序列为:5'-GCCGACACAGCCAGGCCAGGAGG-3'(最后3 个碱基为PAM 序列)。为了将sgRNA 序列克隆至pX333 载体Bbs I 酶切位点,在sgRNA 序列两端添加黏性末端,并合成以下寡脱氧核苷酸序列:sgRNA-F:5'-CACCGCCGACACAGCCAGGCCAGG-3';sgRNA-R:5'-AAACCCTGGCCTG GCTGTGTCGGC-3'。
1.2.2 pX333-Pmepa1 质粒的 构建将sgRNA-F 和sgRNA-R 单链寡脱氧核苷酸在T4 连接酶缓冲液中按照5℃/min 的速率从95℃梯度退火至25℃,从而形成双链寡聚脱氧核苷酸,再加入PNK 磷酸化酶于37℃孵育30 min 进行双链寡脱氧核苷酸5'末端磷酸化修饰。然后通过T4 DNA 连接酶将双链寡聚脱氧核苷酸克隆至Bbs I 内切酶线性化的pX333 载体,重组载体转化入E.coli DH5α 感受态细菌中,于含有氨苄青霉素抗性琼脂固体培养基中筛选。第2 天,挑取琼脂平板中数个单克隆菌落进行扩增培养、质粒提取和测序,对于测序正确的质粒(pX333-Pmepa1)进行大量扩增,制备质粒用于下游细胞转染。
1.2.3 TCMK1 细胞培养与pX333-Pmepa1 质粒转染 TCMK1 细胞培养在补充有10%胎牛血清的DMEM 高糖培养基中,培养条件为37℃、5% CO2和100%湿度。待细胞密度生长至90%汇合度时消化接种至6 孔板,接种量1×106个/孔,当细胞达到80%汇合度时,采用阳离子转染试剂聚乙烯亚胺(PEI)转染500 ng pX333-Pmepa1,转染后6 h 更换新鲜培养基,并在48 h 后于荧光显微镜下观察载体荧光蛋白标签mCherry 的表达,以判断转染是否成功。
1.2.4 流式分选、单克隆细胞扩增和测序鉴定Pmepa1 敲除细胞 将转染后的细胞采用流式分选技术筛选出转染成功的TCMK1 细胞,按照0.5 个细胞/孔的接种比例接种于96 孔板进行5-10 d 培养,每5 d 更换新鲜培养基。将96 孔板中成功生长的单克隆细胞消化,并分别接种于24 孔板,待24 孔板的细胞长满,继续消化并分别接种于6 孔板,待6孔板细胞长满,将其消化下来,一半细胞冻存,一半细胞采用基因组提取试剂盒提取DNA,采用PCR扩增Pmepa1 敲除位点附近基因组片段,PCR 产物送生工生物工程(上海)进行测序分析。若Pmepa1靶点出现序列突变,且两个等位基因均出现移码突变(缺失或插入碱基数非3 的倍数)时,即判定为潜在的表达缺失突变。PCR 鉴定引物为:F,5'-GTTCGTGCAAATCGTGGTCA-3';R,5'-TCATACCTG ACACTTCCTGC-3'。
1.2.5 Western blot 检测Pmepa1 敲除细胞Pmepa1 蛋白的表达 正常TCMK1 细胞和上述Pmepa1 突变TCMK1 细胞分别种于6 孔板,待细胞长满,弃去培养基,PBS 清洗,加入100 μL RIPA 裂解液,静置5 min,充分裂解,用细胞刮将细胞刮下,超声后冰上静置30 min,离心取上清,测定蛋白浓度。向10%的聚丙烯酰胺凝胶中加入各组蛋白样品进行电泳分离,并转印至PVDF 膜上。印迹膜采用5%脱脂牛奶室温封闭1 h,然后,采用含兔抗Pmepa1 抗体(1∶1 000 稀释于2.5% BSA 中)4℃孵育过夜。第2 天,除去一抗,TBST 溶液清洗3 次,每次5 min,加入稀释后的HRP 标记羊抗兔二抗,室温孵育1 h,TBST 洗膜3 次,每次5 min,采用超敏ECL 化学发光底物曝光并上机检测。
1.2.6 PCR、Western blot 检测Pmepa1 敲除后TGF-β1诱导的R-Smad 活化和纤维化表型 将Pmepa1 敲除细胞和正常的TCMK1 细胞接种于6 孔板,培养至细胞汇合度至90%,更换0.5%胎牛血清培养基,饥饿6 h,加入5 ng/mL TGF-β1 刺激 24 h,Trizol 法提取细胞总RNA,取1 μg RNA 逆转录为cDNA,采用RT-PCR 检测 Collagen I 和 Fibronectin 的表达。扩增引物为 Collagen I: F,5'-ATCCAACGAGATCGAGCTCA-3';R,5'-AAGGGAGCCACATCGATGAT-3',Fibronectin: F,5'-ATGTGGA CCCCTCCTGATAGT -3';R,5'-GCCCAGTGATTTCAGCAAAGG -3'。内参引物为GAPDH: F,5'-GGTGAAGGTCGGTGTGAACG -3';R,5'-CTCGCTCCTGGAAGATGGTG -3'。将Pmepa1 敲除细胞和正常的TCMK1 细胞接种于6 孔板,培养至细胞汇合度至90%,更换含0.5% 胎牛血清的培养基饥饿6 h,然后加入5 ng/mL TGF-β1 刺激30 min,提取细胞蛋白。如上采用Western blot 实验检测Smad2、p-Smad2、Smad3、p-Smad3 的表 达,采用GAPDH 为内参。
1.2.7 统计学分析 采用SPSS23.0 或GraphPad Prism8.0 软件进行统计分析,所有的数据结果均表示为均数±标准差(SD),两组间比较采用Student’s t-test 检验,多组间比较采用单因素方差分析(one-way,ANOVA),以P<0.05 表示差异有统计学意义。
2 结果
2.1 成功构建靶向Pmepa1基因CRISPR/Cas9敲除质粒
根据spCas9 G(N19)NGG 的序列识别原则,sgRNA 序列选择在小鼠Pmepa1 基因组第2 个外显子上(图1-A),序列为5'-GCCGACACAGCCAGGCCAGG -3',邻近PAM 序列为5'-AGG -3',该序列经过NCBI BLAST 软件比对分析,确证在小鼠基因组范围内不存在其他非靶向性位点。构建好的载体(pX333-Pmepa1)经测序显示,sgRNA 成功克隆到pX333 载体gRNA 骨架序列5'端,U6 启动子完整驱动gRNA 序列的表达以及CAG 启动子驱动的spCas9 蛋白和mCherry 荧光示踪蛋白同时表达,表明sgRNA 序列克隆成功(图1-B)。
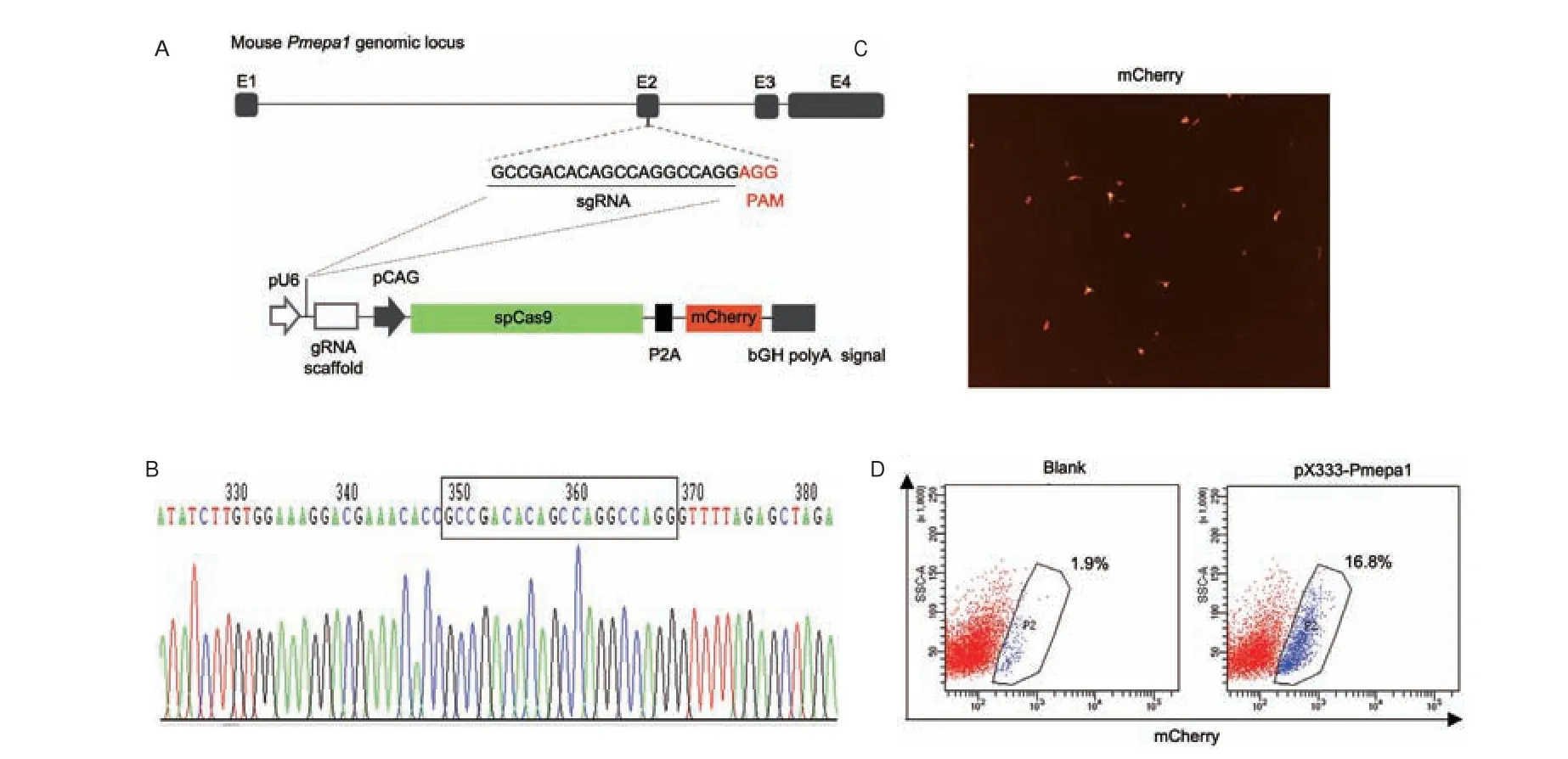
图1 Pmepa1 sgRNA 靶向位置和质粒构建结果Fig.1 Pmepa1 sgRNA targeting location and plasmid construction results
2.2 Pmepa1敲除TCMK1细胞系的构建和鉴定
将构建好的pX333-Pmepa1 质粒转染入TCMK1细胞48 h 后,如图1-B 所示,可以观察到转染成功细胞出现mCherry 荧光蛋白表达(图1-C),流式细胞术显示转染效率约为17%(图1-D)。进一步,采用流式分选技术分析出转染成功(mCherry 阳性)的TCMK1 细胞,按照0.5 细胞/孔的比例稀释接种于96 孔板中进行单克隆细胞培养扩增,最终获得19 个细胞克隆。对这些细胞克隆基因组Pmepa1 位点Cas9 靶向位点序列进行PCR 扩增,最终16 个细胞克隆PCR 扩增成功(图2-A)。经测序分析发现,16 个细胞克隆中2 个克隆的Pmepa1 基因Cas9 靶向位点出现序列突变。且两个突变克隆中序列突变在Pmepa1 两个等位基因位点上序列一致,均为纯合突变。其中clone 4 插入突变1 个碱基,clone 16 缺失突变74 个碱基(图2-B)。由于两个突变细胞克隆缺失/插入碱基数不是3 的倍数,预计会出现开放式阅读框移位,引起后续多肽序列的紊乱和/或蛋白翻译提前终止,从而导致Pmepa1 蛋白序列和功能的缺失。进一步,Western blot 分析发现,clone 4和16 Pmepa1 蛋白表达缺失(图2-C)。这些结果提示,TCMK1 细胞克隆clone 4(KO-4)和16(KO-16)Pmepa1 基因敲除成功。
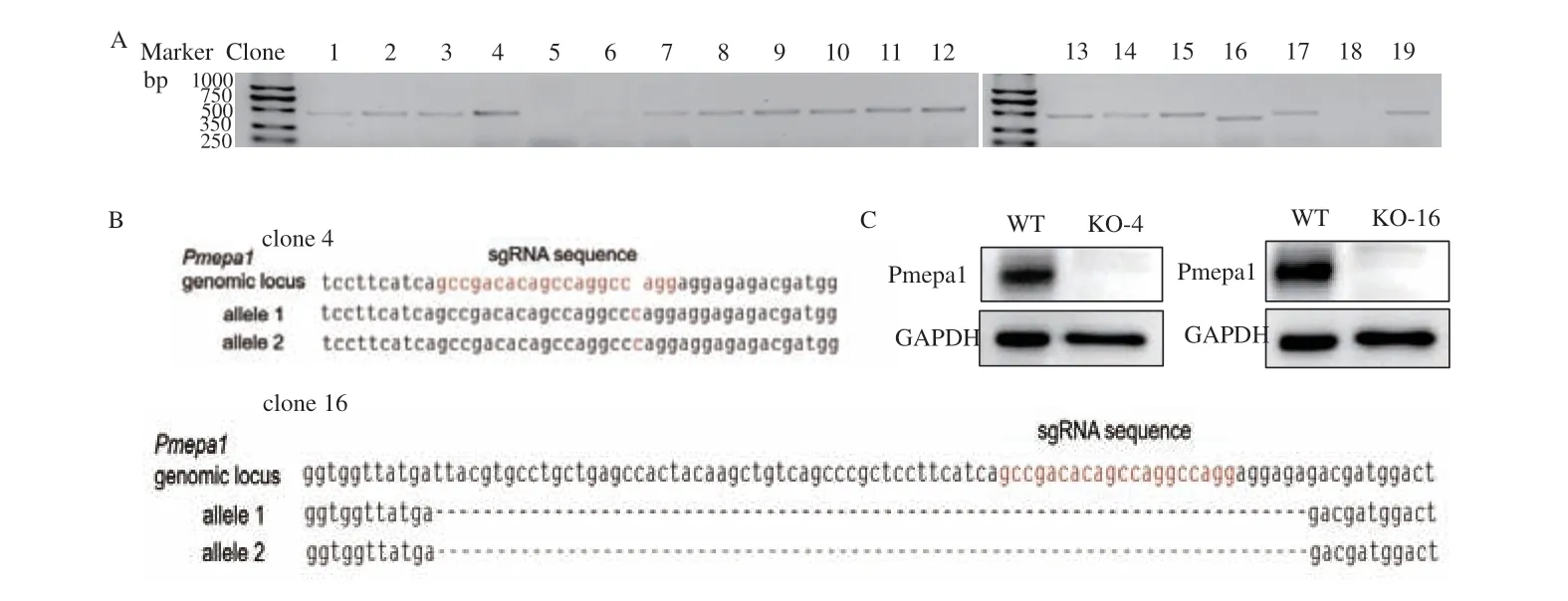
图2 Pmepa1 基因敲除效果验证Fig.2 Validation of Pmepa1 gene knockout
2.3 Pmepa1敲除后TCMK1细胞中TGF-β信号的活化和纤维化
为了研究Pmepa1 敲除是否影响TCMK1 细胞TGF-β 信号的活性,我们采用RT-PCR 及Western blot 方法检测了纤维化相关蛋白及Smad2/Smad3通路相关蛋白。RT-PCR 结果显示,虽然TGF-β1均可以刺激正常和敲除(KO-4/16)TCMK1 细胞Fibronectin 和Collagen I 的表达,但Pmepa1 敲除细胞中Fibronectin 和Collagen I 表达水平显著高于正常TCMK1 细胞,差异均有统计学意义(图3-A)。同时,Western blot 结果显示,TGF-β1 可以刺激TCMK1 细胞中Smad2/3 的活化(磷酸化),但敲除Pmepa1 细胞中(KO-4/16)TGF-β1 诱导的Smad2/3 磷酸化水平更高(图3-B)。因此,PCR 及Western blot 结果表明,Pmepa1 的敲除促进了TCMK1 细胞中TGF-β信号活化和纤维化。
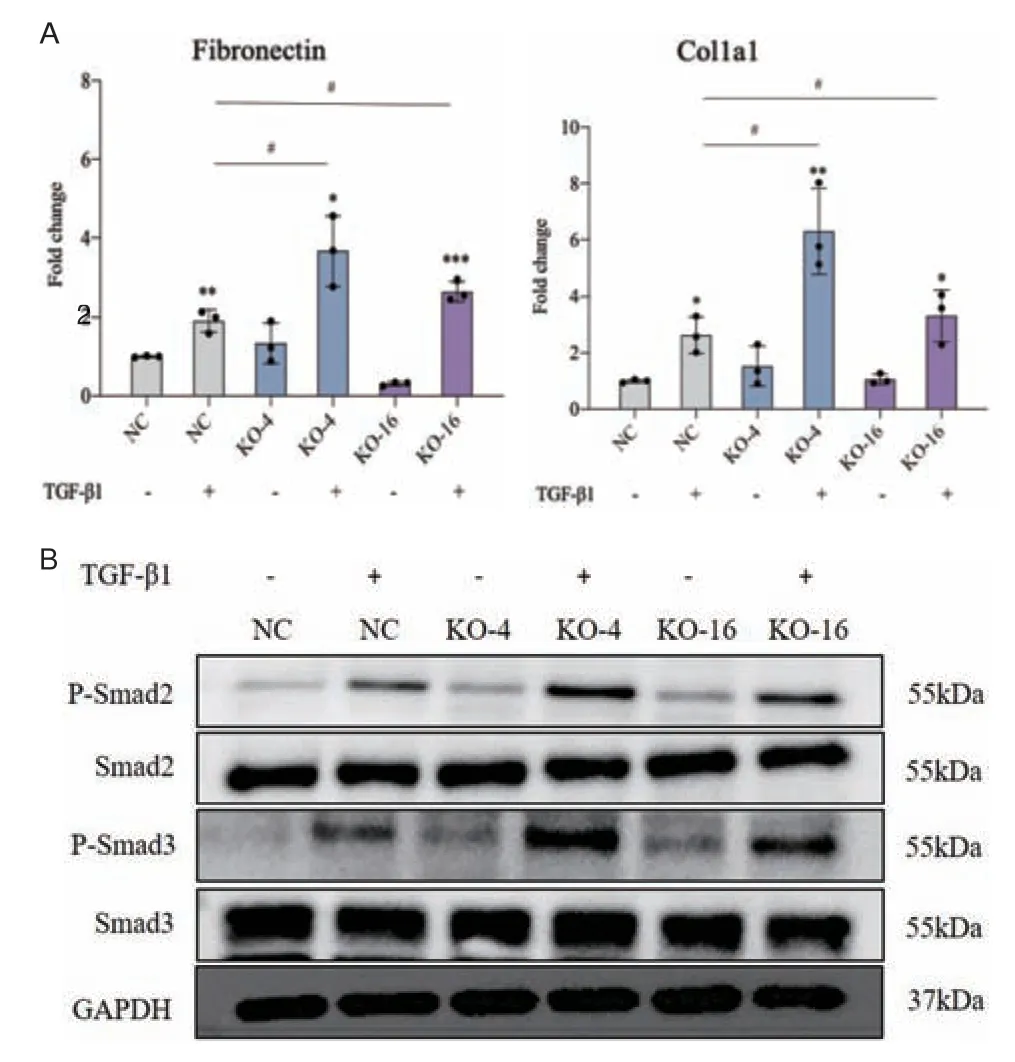
图3 RT-PCR 和Western blot 验证Pmepa1 敲除后TGF-β信号的活化和纤维化蛋白表达结果Fig.3 RT-PCR and Western blot validation of TGF-β signaling activation and fibrotic protein expression after Pmepa1 knockdown
3 讨论
CRISPR/Cas9 技术利用sgRNA 对基因组靶位点序列的识别引导Cas9 核酸酶进行特异性DNA 剪切实现基因敲除[17]。本研究利用CRISPR/Cas9 基因编辑技术敲除TCMK1 细胞Pmepa1 基因,相比其他传统基因编辑技术,CRISPR/Cas9 基因编辑技术具有设计简单、周期快、高切割率、高精度的特点[18]。实验表明CRISPR/Cas9 技术在小鼠上建立基因敲除模型最快6-8 周,而且成功率较高[19]。陈汉宗等[20]成功利用CRISPR/Cas9 技术稳定构建了anxa6 基因敲除的Caco-2 细胞株。刘铁柱等[21]建立了SNX11基因稳定敲除的A549 细胞系。CRISPR/Cas9 基因编辑技术目前已被广泛应用于基因的筛选、动物基因的敲入等实验以及各遗传疾病的治疗中[22]。
Pmepa1 又称TMEPA1,在TMEPA1 家族分子中发现,C18ORF1 与TMEPA1 一样具有两个PY基序和一个Smad 相互作用基序(SIM)结构域,C18ORF1 可以阻断TGF-β 信号传导,但不能阻断骨形态发生蛋白信号传导[23]。C18ORF1 通过SIM与Smad2/3 结合,并与Smad2/3 的受体激活,以减弱Smad2/3 向TGF-I 型受体的募集,其对TGF-β信号传导的抑制功能与TMEPAI 方式相似[24]。但C18ORF1 与TMEPAI 相反的是,TGF-β 信号传导没有诱导C18ORF1,而当细胞受到高水平的TGF-β刺激时,TMEPAI 可能会帮助C18ORF1 以协调的方式 抑制TGF-β 信号传导[25]。Pmepa1 表达 在细胞膜和亚细胞细胞器中,其表达可以被雄激素、TGF-β、EGF 和Wnt 等多种细胞内信号通路诱导[26]。Pmepa1 基因首次发现于前列腺癌症中,在前列腺癌症中具有双重作用。Pmepa1 可以促进雄激素受体阳性前列腺癌细胞增生,并且通过抑制Smad3/4 来减少雄激素受体阴性前列腺癌细胞的增生[27]。此外,Pmepa1 可以促进其他癌症的发展,如肺癌、卵巢癌、胃癌等[28]。实验证明,Pmepa1 在卵巢癌症中的抗癌作用可以促进凋亡[29]。这些差异可能由组织的特异性引起的。Pmepa1 的不同作用可以通过Pmepa1和TGF-β 的关系来解释。Pmepa1 是由TGF-β 信号传导诱导的,是TGF-β 信号传导的直接靶基因。但同时,它抑制典型的Smad 信号传导,实验证明Pmepa1 通过抑制Smad2 和Smad3 的磷酸化以对抗TGF-β 信号传导[30]。TGF-β1 是组织修复、炎症和纤维化的主要调节因子,也有大量的实验数据表明肾纤维化与TGF-β 有关[31]。考虑到Pmepa1 和TGF-β 信号传导的关系,我们为此验证了Pmepa1 在TGF-β 信号活化的TCMK1 细胞中是否发挥抗纤维化的作用。
本实验利用CRISPR/Cas9 基因编辑技术联合流式细胞分选,成功建立了2 株Pmepa1 基因敲除TCMK1 细胞系,敲除细胞经过Western blot 验证了Pmepa1 蛋白表达的缺失,单细胞克隆筛选成功率约10%。为明确Pmepa1 基因的敲除与TCMK1 细胞中TGF-β 信号的活化和纤维化的关系,通过RT-PCR及Western blot 两种方法检测了纤维化相关蛋白及Smad2/Smad3 通路相关蛋白,结果表明Pmepa1 的敲除促进了TCMK1 细胞中TGF-β 信号活化和纤维化。
4 结论
本实验通过CRISPR/Cas9 基因编辑技术成功构建了Pmepa1 敲除的TCMK1 细胞系,并且针对两株敲除细胞系通过RT-PCR 及Western blot 两种方法验证 了Pmepa1 敲除后TCMK1 细胞中TGF-β 信号的活化和纤维化,结果显示Pmepa1 的敲除促进了TCMK1 细胞中TGF-β 信号活化和纤维化。不仅为Pmepa1 蛋白在TCMK1 小鼠肾小管上皮细胞中的作用以及在肾纤维化中提供细胞模型,也为后续进一步研究肾脏组织中Pmepa1 基因与肾脏疾病的关系奠定基础。
