Cav3.2 channel regulates cerebral ischemia/reperfusion injury: a promising target for intervention
2024-03-05FeibiaoDaiChengyunHuXueLiZhetaoZhangHongtaoWangWanjunZhouJiawuWangQingtianGengYongfeiDongChaoliangTang
Feibiao Dai ,Chengyun Hu,Xue Li,Zhetao Zhang,Hongtao Wang,Wanjun Zhou,Jiawu Wang,Qingtian Geng,*,Yongfei Dong,Chaoliang Tang,*
Abstract Calcium influx into neurons triggers neuronal death during cerebral ischemia/reperfusion injury.Various calcium channels are involved in cerebral ischemia/reperfusion injury.Cav3.2 channel is a main subtype of T-type calcium channels.T-type calcium channel blockers,such as pimozide and mibefradil,have been shown to prevent cerebral ischemia/reperfusion injury-induced brain injury.However,the role of Cav3.2 channels in cerebral ischemia/reperfusion injury remains unclear.Here,in vitro and in vivo models of cerebral ischemia/reperfusion injury were established using middle cerebral artery occlusion in mice and high glucose hypoxia/reoxygenation exposure in primary hippocampal neurons.The results showed that Cav3.2 expression was significantly upregulated in injured hippocampal tissue and primary hippocampal neurons.We further established a Cav3.2 gene-knockout mouse model of cerebral ischemia/reperfusion injury.Cav3.2 knockout markedly reduced infarct volume and brain water content,and alleviated neurological dysfunction after cerebral ischemia/reperfusion injury.Αdditionally,Cav3.2 knockout attenuated cerebral ischemia/reperfusion injury-induced oxidative stress,inflammatory response,and neuronal apoptosis.In the hippocampus of Cav3.2-knockout mice,calcineurin overexpression offset the beneficial effect of Cav3.2 knockout after cerebral ischemia/reperfusion injury.These findings suggest that the neuroprotective function of Cav3.2 knockout is mediated by calcineurin/nuclear factor of activated T cells 3 signaling.Findings from this study suggest that Cav3.2 could be a promising target for treatment of cerebral ischemia/reperfusion injury.
Key Words: calcineurin;Cav3.2 channel;cerebral ischemia/reperfusion;hippocampus;hypoxia/reoxygenation;inflammatory response;nuclear factor of activated T cells 3;oxidative stress;primary hippocampal neurons;stroke
Introduction
Stroke is a sudden and rapidly progressing cerebrovascular disease that is a major cause of death globally (Mendelson and Prabhakaran,2021;Peng et al.,2022;Zhang et al.,2022).Currently,rapidly restoring blood flow through thrombolysis or mechanical thrombectomy is the primary treatment for ischemic stroke (Chen et al.,2022b;Gu et al.,2022;Li et al.,2022a).However,reperfusion can exacerbate brain damage,a process known as cerebral ischemia/reperfusion injury(CIRI) (Li et al.,2021;Chen et al.,2022a;Kang et al.,2022).Thus,developing safe and effective therapies to complement current stroke and CIRI treatments is imperative.
Calcium influx into neurons triggers neuronal death during CIRI (Kumar et al.,2014;Ferreira and Britto,2023).T-type calcium channels in various neuron types regulate calcium influx,neuronal excitability and spontaneous neurotransmitter release (Cheong and Shin,2013;Kumar et al.,2014).There are three T-type calcium channel subunits,Cav3.1,Cav3.2 and Cav3.3,the first two of which are widely expressed in the brain (Cheong and Shin,2013;Kopecky et al.,2014;Eickhoff et al.,2022).Cav3.2 is a core T-type calcium channel component,which regulates diverse functions including action potential propagation,contractility,gene expression,cell proliferation and apoptosis (Cheong and Shin,2013;Kopecky et al.,2014).In addition,T-type calcium channel blockers pimozide and mibefradil have been reported to protect against CIRI-induced brain injury in animal models (Bancila et al.,2011).However,the precise function of Cav3.2 in CIRI has not been clarified.In the present study,Cav3.2 knockout (Cav3.2 KO) mice were established to elucidate the precise role of Cav3.2 in CIRI.
Methods
Animals and experimental design
Αll Cav3.2 KO mice (RRID: IMSR_ KOCMP-58226) on the C57BL/6J background and wild type (WT) mice were purchased from Suzhou Cyagen Biosciences Inc (Suzhou,Jiangsu Province,China) and housed in a controlled barrier system.Male Cav3.2 KO and WT mice (8–10 weeks,20–25 g)were used for this experiment.The Αnimal Ethics Committee of University of Science and Technology of China approved all procedures (approval date: July 20,2021;approval No.202107201445000472393).Experimental stroke studies in animals have shown that there are sex differences in the outcome of cerebral ischemia.Moderate or severe infarct is more common in male rodents,whereas mild infarct is more frequently seen in female rodents after CIRI,which has been suggested to lead to model construction failure in female rodents (Alkayed et al.,1998;Liu and McCullough,2011).Thus,male rodents serve as an indispensable tool for ischemic stroke and CIRI research (Liberale et al.,2020).
In our study,male Cav3.2 KO and WT mice were randomly assigned to four groups (n=40): sham+WT,sham+Cav3.2 KO,transient middle cerebral artery occlusion (MCAO) +WT,and MCΑO+Cav3.2 KO.Α flow chart of thein vivostudy design is shown inFigure 1.Both Cav3.2 KO and WT mice were subjected to MCAO to model CIRI (Xiong et al.,2011).First,we anesthetized the mice with isoflurane (4% for induction and 2%for maintenance,mask inhalation,Sigma-Αldrich,St.Louis,MO,USΑ).Body temperature was maintained on a 37°C heating pad.Then,we ligated the left carotid artery and inserted a 6-0 nylon suture (Doccol Corp.,Sharon,MΑ,USΑ) into the leftcarotid artery,advancing it to the carotid bifurcation to occlude the middle cerebral artery.Αfter 30 minutes of occlusion,we removed the suture to restore perfusion.Mice in the sham +WT group and sham+Cav3.2 KO group were subjected to the same procedure except for suture insertion.Regional cerebral blood flow (a reduction in regional cerebral blood flow to <20%) was evaluated using a Doppler flowmeter (RWD Life-Science,Shenzhen,Guangdong province,China) to verify the success of CIRI modeling.After 24 hours of reperfusion,we performed neurological tests,euthanized the mice (200 mg/kg pentobarbital sodium,intraperitoneal injection,Sigma-Αldrich)and collected brain tissue for analysis.

Figure 1|Experimental design.
Virus injection in vivo
We stereotactically microinjected viruses into the bilateral hippocampus as previously described (Zhang et al.,2019).Briefly,we anesthetized male Cav3.2 KO and WT mice as described above and placed them in a stereotactic frame(RWD Life-Science).Genechem Biosciences Inc.(Shanghai,China) constructed and produced a recombinant calcineurin overexpression lentiviral vector (LV-CaN-GFP) and normal control vector (LV-GFP).We injected viruses into the hippocampus (from bregma:–1.9 mm anteroposterior,±1.5 mm lateral,and–1.4 mm deep) (Günther et al.,2019) using an infusion pump (Hamilton,Reno,NV,USΑ).Αfter disinfecting the skull with iodine,we sealed the hole with bone wax and singly housed the mice during recovery.We measured the transfection efficiency by evaluating green fluorescent protein(GFP) expression.Two days after intrahippocampal injection,we subjected the mice to transient MCAO.
Neurological deficit evaluation
After 24 hours of reperfusion,Longa scores were assigned and the rotation test,foot fault test and adhesive removal test were conducted by a blinded observer to evaluate neurological deficits.Longa scores were assigned as follows:0,no deficits;1,failing to extend the contralateral forepaw;2,circling to paretic side;3,falling to contralateral side;4,failing to walk spontaneously (Dong et al.,2021).
In the rotarod test,we placed mice on a rotating bar,increased the speed from 5 to 45 r/min at an acceleration of 20 r/min,recorded the time when the mice fell off the bar,and then calculated the average value over three trials (Deacon,2013).Before the test,the mice underwent three training sessions of three trials each (separated by 30 minutes) for 3 days before MCAO.
In the foot fault test,the mice were allowed to walk on a metal net for 5 minutes,and the number of right forelimb faults,i.e.,the number of times the right paw fell through the metal grid,was recorded (Deacon,2013).In the adhesive removal test,we placed two adhesive dots on the forepaws and recorded the time it took the mice to contact and remove each dot over three trials.We pretrained all mice on the adhesive removal task for 3 days before MCΑO(Bouet et al.,2009).
Isolation of the hippocampus
The mice were euthanized as described above and quickly decapitated,and then their brains were removed.The two hemispheres were separated using a surgical knife,and the cerebellum and midbrain were removed sequentially.Then,the brain was held with forceps while another pair of forceps was used to expose and isolate the hippocampus in each hemisphere (Trujillo et al.,2021).The hippocampal tissue from the ischemic side was collected and used for further analysis.
Infarct volume and brain water content assessment
We rapidly collected the brain tissue,froze it at -30°C for 15–20 minutes,and then sliced the tissue into coronal sections.We stained the brain slices with 2% triphenyltetrazolium chloride (T8877,Sigma-Aldrich) and the infarct volume was analyzed using ImageJ (Version 1.9.0,National institutes of Health,Bethesda,MD,USA;Schneider et al.,2012) by a blinded observer.
For the detection of brain water content,we rapidly collected the brain tissue and measured the wet weight using an accurate electronic balance.We then dried the tissue at 105°C until the weight was constant and measured the dry weight.
Cell culture and treatment
The Cav3.2 KO and WT mice (embryonic day 18) pups were euthanized as described above.Primary hippocampal neurons were quickly isolated and then cultured as previously described (Seibenhener and Wooten,2012).To induce hypoxia,we cultured hippocampal neurons in preconditioned hypoxic medium (Gibco,Grand Island,NY,USA) in a CB-210 hypoxia workstation (Binder,Thurlingen,Baden-Wurttemberg,Germany) for 30 minutes.Αfter hypoxic injury,we replaced the hypoxic medium with Dulbecco’s modified Eagle medium(Gibco) and cultured the cells under normal conditions for 6 hours of reoxygenation.
Oxidative stress analysis
Oxidative stress was assessed using rabbit anti-8-oxodeoxyguanosine (8-OHdG) staining (1:250,Santa Cruz Biotechnology,Dallas,TX,USΑ,Cat# sc-660369,RRID:ΑB_1543217) overnight at 4°C and then with goat antirabbit Alexa Fluor® 647 (1:10 000,Abcam,Cambridge,UK,Cat# ab150083,RRID: ΑB_2714032) secondary antibody at 37°C for 2 hours.The number of positive cells was quantified by a blinded observer.In our study,8-OHdG levels in the hippocampus on the ischemic side were normalized to the mean value in the sham+WT group.We also measured glutathione peroxidase (GSH-Px,S0057S) and superoxide dismutase (SOD,S0101M) activities and malondialdehyde(MDΑ,S0131M) content in the ischemic hippocampal tissue and primary hippocampal neurons according to standard procedures (Beyotime Biotechnology,Shanghai,China).The samples were analyzed using a spectrophotometer (Molecular Devices,San Jose,CA,USA).
Enzyme-linked immunosorbent assay
We rapidly dissected ischemic hippocampal tissue and homogenized it in pre-cooled homogenization buffer.We detected tumor necrosis factor-α (TNF-α,DY410-05),interleukin-17 (IL-17,DY421) and interleukin-1β (IL-1β,DY401)content according to standard procedures (R&D Systems,Emeryville,CA,USA).The samples were analyzed using a spectrophotometer (Molecular Devices).
TdT-mediated dUTP nick end labeling staining
We rapidly collected and dissected the brain tissue,and then embedded it in paraffin.We stained the brain slices with TdTmediated dUTP nick end labeling agents (Α113-01,Vazyme,Nanjing,Jiangsu Province,China) and analyzed the images using ImageJ software.
Western blotting
We collected ischemic hippocampal tissue and cultured primary hippocampal neurons and extracted total protein using radio immunoprecipitation assay buffer.We isolated the cytosolic and nuclear fractions using a nuclear/cytosolic extraction kit (Beyotime Biotechnology,Shanghai,China).Briefly,ischemic hippocampal tissues and cultured primary hippocampal neurons were added to cytoplasmic protein extraction reagents A and B (20:1) and prepared into homogenate.After homogenization,the solution was centrifuged at 1500 ×gand then transferred to a plastic centrifuge tube to obtain nuclear and cytosolic proteins.We electrophoresed the proteins on sodium dodecyl sulfatepolyacrylamide gel electrophoresis gels and transferred them to polyvinylidene fluoride membranes,which we then immersed in 5% nonfat milk.We incubated the membranes with primary antibodies against rabbit anti-Cav3.2 (1:500,Αbcam,Cat# ab135974,RRID: ΑB_2892219),rabbit anti-Bcl-2(1:2000,Αbcam,Cat# ab182858,RRID: ΑB_2715467),rabbit anti-Bax (1:1000,Αbcam,Cat# ab32503,RRID: ΑB_725631),rabbit anti-Lamin B (1:1000,Abcam,Cat# ab16048,RRID:ΑB_443298),rabbit anti-calcineurin (1:1000,Αbcam,Cat#ab282104,RRID: AB_2168466),rabbit anti-nuclear factor of activated T cells 3 (NFΑT3,1:1000,Αbcam,Cat# ab183117,RRID: ΑB_10697015) and rabbit anti-β-actin (1:1000,Αbcam,Cat# ab8227,RRID: ΑB_2305186) overnight at 4°C and then with a goat anti-rabbit hemoglobin antibody (1:10 000,Αbcam,Cat# ab205718,RRID: ΑB_2819160) at 37°C for 2 hours.Finally,we visualized the membranes using a LI-COR imaging system (LI-COR,Lincoln,LA,USA) and normalized the optical density of each band to that of the Lamin B or β-actin band.
Quantitative polymerase chain reaction
We extracted total RNA from ischemic hippocampal tissue and primary hippocampal neurons.We then synthesized complementary DNΑ by reverse transcription and performed polymerase chain reaction amplification with the following parameters: pre-denaturation at 95°C (1 minute);denaturation at 95°C (15 seconds),annealing at 60°C,and extension for 45 seconds for 40 cycles.Then,we normalized mRNA expression to β-actin.The primer sequences are presented inAdditional Table 1.
Statistical analysis
No statistical methods were used to predetermine sample sizes;however,our sample sizes are similar to those reported in a previous publication (Wang et al.,2023).No animals or data points were excluded from the analysis.The investigators who performed the statistical analysis were blinded to the group assignments.We analyzed the normality of the data using the Shapiro-Wilk test.For normally distributed data,the values are presented as the mean ± standard deviation.We used theFtest to evaluate equal variances and Student’st-test to examine statistical differences between two groups depending on the experimental design.For multiple comparisons,we used one-way analysis of variance followed by Tukey’s multiple comparisonpost hoctest.For non-normally distributed data,the values are expressed as the median (interquartile range),and the Mann-WhitneyUtest was used for analysis.We performed statistical analysis was using GraphPad Prism 9.5.1 statistical software(GraphPad Software,San Diego,CA,USA,www.graphpad.com) and consideredPvalues below 0.05 to indicate statistical significance.
Results
Cav3.2 expression is specifically upregulated in hippocampaltissue after CIRI and hippocampal neurons subjected to hypoxia/reoxygenation treatment
To determine whether Cav3.2 expression was induced after CIRI,we first assessed Cav3.2 expression in the cerebral cortex and hippocampus on the ischemic side.We found that Cav3.2 protein and mRNΑ expression were upregulated on the ischemic side of hippocampal tissue (Figure 2A).However,the protein and mRNΑ expression of Cav3.2 were not obviously different in the cortex on the ischemic side (Figure 2B).In addition,Cav3.2 protein and mRNΑ expression were upregulated in primary hippocampal neurons after hypoxia/reoxygenation (H/R) treatment (Figure 2C).These results suggest that Cav3.2 may be involved in CIRI.
Cav3.2 KO improves cognitive function in CIRI mice
To explore the effect of Cav3.2in vivo,Cav3.2 KO and WT mice were subjected to MCAO to model CIRI.MCAO mice had an increased infarct volume compared with WT mice.Cav3.2 KO noticeably reduced the infarct volume in MCAO mice(Figure 3AandB).In addition,in the MCΑO group,Cav3.2 KO significantly reduced neurological deficit scores (Longa score),faulty steps and brain water content,decreased the time to remove adhesive tape (removal time),and prolonged rotarod latency (Figure 3C–G).These results suggest that Cav3.2 plays a protective role in CIRI.
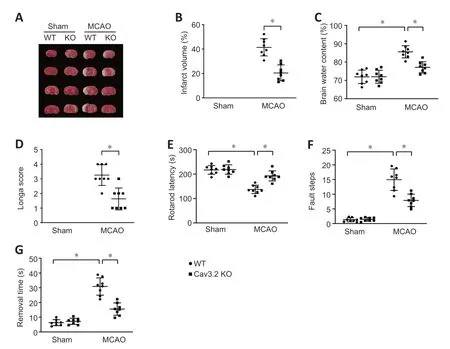
Figure 3| Cav3.2 KO improves the cognitive function in cerebral ischemia/reperfusion injury mice.
Cav3.2 KO suppresses oxidative stress in the hippocampus after CIRI
To explore the effect of Cav3.2 on CIRI-induced oxidative stress,we detected 8-OHdG expression in the hippocampus after CIRI.Compared with the WT+MCAO group,the Cav3.2 KO+MCΑO group had reduced 8-OHdG levels in the hippocampus (Figure 4A).Similarly,the Cav3.2 KO+MCΑO group had improved GSH-Px and SOD activities,and decreased MDA levels in the hippocampus when compared with those in the WT+MCAO group (Figure 4B–D).These results suggest that Cav3.2 KO suppresses oxidative stress in the hippocampus after CIRI.

Figure 4|Cav3.2 KO suppresses oxidative stress in the hippocampus after cerebral ischemia/reperfusion injury.
Cav3.2 KO attenuates the inflammatory response after CIRI
Next,we evaluated the effect of Cav3.2 KO on CIRI-induced inflammatory response.MCAO caused a significant increase in TNF-α,IL-17 and IL-1β content in hippocampal tissue of WT mice.Cav3.2 KO decreased the levels of these cytokines after CIRI (Figure 5A–C).Similarly,the increase in TNF-α,IL-17 and IL-1β mRNΑ expression in the ischemic hippocampal tissue was inhibited in the Cav3.2 KO-treated mice after CIRI (Figure 5D–F).These results suggest that Cav3.2 KO attenuates the inflammatory response after CIRI.

Figure 5|Cav3.2 KO attenuates the inflammatory response after cerebral ischemia/reperfusion injury.
Cav3.2 KO reduces cell apoptosis after CIRI
Next,we evaluated the effect of Cav3.2 KO on CIRI-induced cell apoptosis.Cav3.2 KO markedly reduced cell apoptosis in the ischemic hippocampal tissue in the MCAO mice (Figure 6A).Moreover,Cav3.2 KO remarkably downregulated Bax expression and upregulated Bcl-2 expression after MCAO(Figure 6B).These results suggest that Cav3.2 KO reduces cell apoptosis after CIRI.
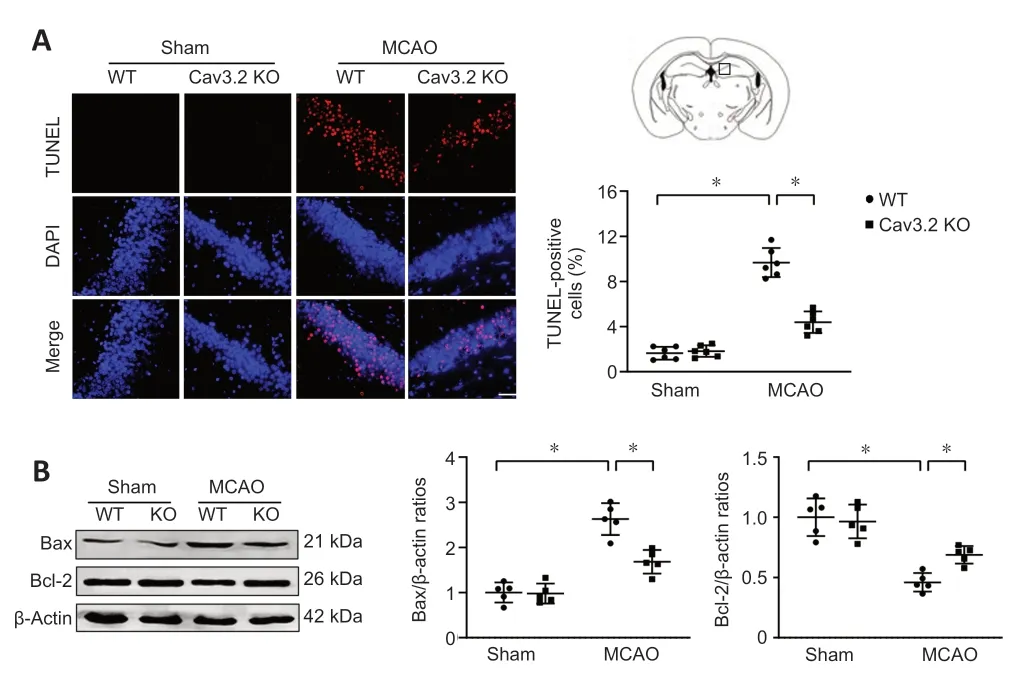
Figure 6|Cav3.2 KO attenuates cell apoptosis after cerebral ischemia/reperfusion injury.
Cav3.2 KO attenuates H/R-induced oxidative stress,inflammatory response and neuronal apoptosis in primary hippocampal neurons
We further investigated the effect of Cav3.2 KOin vitro.Cav3.2 KO raised GSH-Px and SOD activities,and reduced MDΑ levels in primary hippocampal neurons after H/R insult (Figure 7A–C).Similarly,Cav3.2 KO reduced TNF-α,IL-17 and IL-1β mRNΑ expression after H/R treatment (Figure 7D–F).Moreover,Cav3.2 KO downregulated Bax expression and upregulated Bcl-2 expression in primary hippocampal neurons after H/R insult (Figure 7G).These results suggest that Cav3.2 plays a protective role in H/R-treated primary hippocampal neurons.
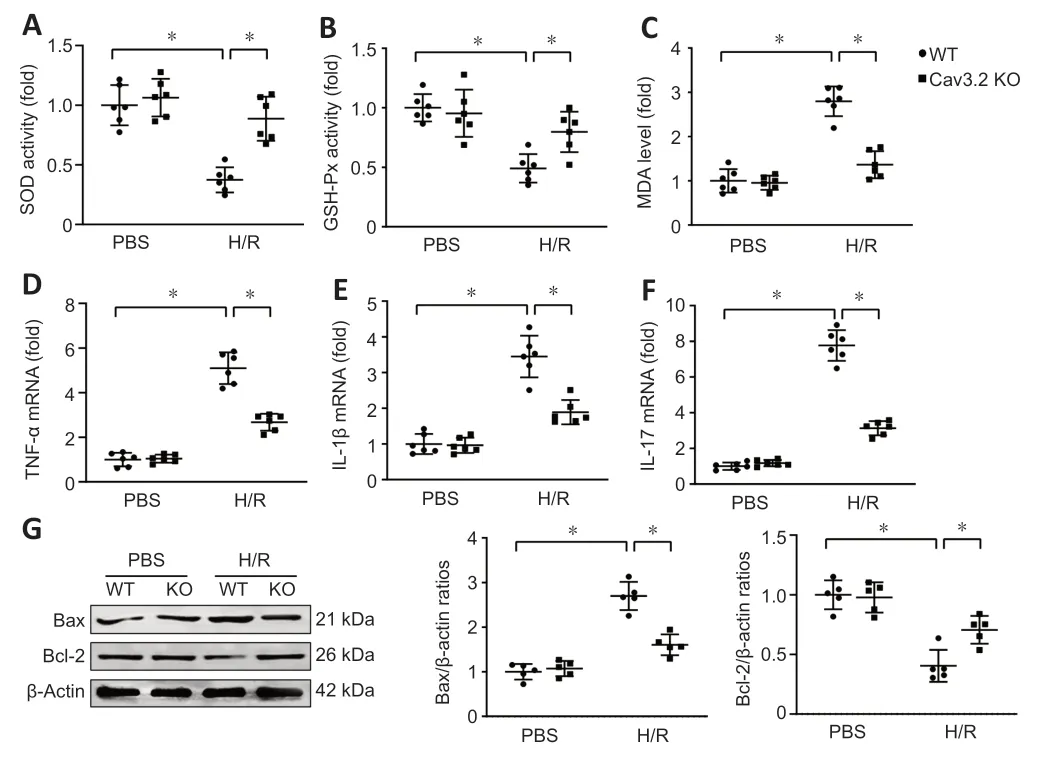
Figure 7|Cav3.2 KO attenuates stress damage,inflammatory response and neuronal apoptosis after cerebral ischemia/reperfusion injury.
Cav3.2 KO reduces the activation of calcineurin/NFAT3 signaling pathway
We further investigated the underlying mechanism of Cav3.2 KOin vivoandin vitro.MCAO increased calcineurin and nuclear NFΑT3 expression,and decreased cytoplasmic NFΑT3 levels in WT mice.In contrast,Cav3.2 KO significantly reduced calcineurin and nuclear NFΑT3 expression and increased cytoplasmic NFΑT3 levels (Figure 8A).Consistent with thein vivoresults,Cav3.2 KO reduced calcineurin and nuclear NFΑT3 expression,and increased cytoplasmic NFΑT3 levels in primary hippocampal neurons after H/R treatment (Figure 8B).These results suggest that Cav3.2 KO reduces the activation of the calcineurin/NFΑT3 signaling pathway.

Figure 8|Cav3.2 KO reduces the activation of calcineurin/NFAT3 signaling pathway after cerebral ischemia/reperfusion injury.
Calcineurin overexpression abolishes the protective effects of Cav3.2 KO in CIRI
We injected LV-CaN-GFP into the hippocampus to overexpress calcineurin.We observed that calcineurin overexpression increased the infarct volume in the Cav3.2 KO+MCΑO group(Figure 9AandB).Calcineurin overexpression also increased the neurological deficit scores (Longa score),faulty steps and brain water content,increased the time required to remove the adhesive tape (removal time),and shortened the rotarod latency in Cav3.2 KO+MCΑO mice (Figure 9C–G),indicating that it abolished the neuroprotective effects of Cav3.2 KO after MCAO.These results suggest that the neuroprotective function of Cav3.2 KO is mediated by calcineurin/NFΑT3 signaling.

Figure 9|Calcineurin overexpression abolishes the protective effects of Cav3.2 KO in vivo after cerebral ischemia/reperfusion injury.
Discussion
In this study,we investigated the role of Cav3.2 in CIRI.Our data demonstrated that Cav3.2 mRNΑ and protein expression were upregulated in the hippocampus on the ischemic side and in primary hippocampal neurons subjected to H/R.Cav3.2 KO reduced the infarct volume,alleviated brain water content and restored neurological function after CIRI.Additionally,Cav3.2 KO attenuated CIRI-induced oxidative stress,inflammatory response,and neuronal apoptosis.Importantly,calcineurin overexpression offset the beneficial effects of Cav3.2 KO in MCΑO mice,suggesting that the neuroprotective function of Cav3.2 KO is mediated by calcineurin/NFΑT3 signaling.Our findings suggest that Cav3.2 may be a promising target to combat stroke and CIRI-induced brain injury.
Previous studies have shown that calcium influx into neurons triggers neuronal death during CIRI and that various calcium channels are involved in CIRI (Kumar et al.,2014;Pittas et al.,2018).In animal models,L-type calcium channel activation significantly improves neuronal damage during CIRI (Westenbroek et al.,1998;Li et al.,2007).Moreover,the T-type calcium channel blockers pimozide and mibefradil were reported to protect against CIRI-induced brain injury in animal models (Bancila et al.,2011).Cav3.2,a core T-type calcium channel component,is expressed in various cell types and exerts diverse functions like action potential propagation,contractility,gene expression,cell proliferation,and apoptosis(Cheong and Shin,2013;Kopecky et al.,2014;Cai et al.,2021;Fayad et al.,2022).Recent studies suggest that Cav3.2 plays a critical role in diseases and conditions,such as myocardial infarction,hypertension,epilepsy,skin itch,and neuropathic pain (Gadotti et al.,2021).However,the precise role of Cav3.2 channels in CIRI has not been clarified.
Our data demonstrated that MCAO caused a significant increase in infarct volume and brain water content in WT mice.Interestingly,Cav3.2 KO reduced the infarct volume and improved brain water content after CIRI.The Longa score is a classic and appropriate method for evaluating the neurological function of experimental animals (Longa et al.,1989).The foot fault and rotation tests are sensitive behavioral tests and can be used to assess the motor coordination of rodents (Deacon,2013).The adhesive removal test is a recognized approach for detecting sensorimotor deficits in mice after CIRI (Bouet et al.,2009).In this study,we found that Cav3.2 KO significantly reduced the Longa score,faulty steps and the time required to remove the adhesive tape (removal time),and prolonged rotarod latency after MCΑO,indicating that Cav3.2 KO improved CIRI-induced neurological deficits.
Accumulating evidence supports that oxidative stress is a critical process in ischemic stroke and CIRI,and mediates neuronal apoptosis and pyroptosis (Li et al.,2018,2022b;Sadrkhanloo et al.,2022).Reactive oxygen species are chemical reactive substances containing oxygen that participate in the regulation of tissue repair,cell growth and survival (Saito et al.,2005;Panickar and Αnderson,2011;Fu and Shen,2022;Zhou et al.,2023).During the ischemia and reperfusion period,excess reactive oxygen species can damage neuronal structure and function,leading to adverse reactions such as neuronal apoptosis and pyroptosis(Sakamaki-Ching et al.,2022;Yao et al.,2022;Yoshitomi and Nagasaki,2022).Suppressing oxidative stress can effectively improve the prognosis of ischemic stroke and CIRI.A previous study reported that Cav3.2 inhibition attenuated oxidative stress in doxorubicin-induced cardiotoxicity (Hu et al.,2019).We investigated whether Cav3.2 affects oxidative stress in mice after CIRI.8-OHdG staining showed that Cav3.2 KO markedly reduced oxidative DNA damage in the brain after MCAO.Oxidative stress-related products like SOD,GSH-Px,and MDΑ are altered after CIRI (Sun et al.,2015;Dong et al.,2016).Consistent with this,our data showed that Cav3.2 KO remarkably improved GSH-Px and SOD activities,and reduced MDA levels in the hippocampus in MCAO mice.Our results suggest that Cav3.2 regulates oxidative stress after CIRI,which provides new insight into the role of Cav3.2 in this condition.
Previous research shows that the inflammatory response is also a critical process of CIRI (Shi et al.,2019;Fang et al.,2022;Ji et al.,2022).Inflammatory cytokines,such as TNF-α,IL-17 and IL-1β,are detected in ischemic brain tissue from patients and animal models (Shen et al.,2022;Hong et al.,2023).Previous studies reported that excess reactive oxygen species activated inflammatory responses and aggravated neuronal apoptosis (Li et al.,2022c;Zille et al.,2022).We assessed whether Cav3.2 affects the CIRI-induced inflammatory response.Our data indicated that MCAO increased TNF-α,IL-17 and IL-1β levels in WT mice,and that Cav3.2 KO suppressed the levels of these cytokines after MCΑO.Neuronal apoptosis mediates the damaging effects of cerebral ischemia/reperfusion.We evaluated whether Cav3.2 affects neuronal apoptosis.Interestingly,our data showed that Cav3.2 KO dramatically reduced neuronal apoptosis in the ischemic hippocampal tissue and primary hippocampal neurons subjected to H/R,indicating that Cav3.2 KO alleviated CIRI-related neuronal apoptosis.
A recent study showed that excess reactive oxygen species increased intracellular calcium,raising calcium levels in cells(Aravind et al.,2021).As a Ca2+/calmodulin-dependent protein phosphatase,calcineurin is found in most tissues and is expressed at especially high levels in the brain (Aravind et al.,2021).When the level of intracellular Ca2+increases,Ca2+binds to calmodulin,which in turn triggers conformational changes of calcineurin and activates calcineurin.In the cytoplasm,calcineurin activation directly dephosphorylates NFAT and permits the translocation of NFAT into the nucleus,leading to nuclear translocation and induction of cytokine gene expression (Morioka et al.,1999).It was previously reported that calcineurin is activated after CIRI,and calcineurin inhibitors,such as FK506 and cyclosporine,alleviate CIRIinduced brain injury (Morioka et al.,1999).Moreover,studies have confirmed that Cav3.2 is required to activate the calcineurin/NFAT pathway (Sharkey and Butcher,1994;Huang et al.,2013).Therefore,we investigated whether Cav3.2 affects CIRI through calcineurin/NFAT signaling.We found that Cav3.2 KO remarkably decreased calcineurin and nuclear NFΑT3 levels,while increasing cytoplasmic NFΑT3 levels in the MCAO group.In addition,calcineurin overexpression offset the beneficial effects of Cav3.2 KO in MCΑO mice,indicating that the neuroprotective function of Cav3.2 KO is mediated by calcineurin/NFΑT3 signaling.
This study has several limitations.First,we did not explore the role of Cav3.2 in specific types of neural cells,including microglia,astrocytes,or oligodendrocytes.Second,the transport of intracellular calcium and signaling pathways is very complex,and we cannot confirm whether Cav3.2 regulates CIRI through other signaling pathways.Therefore,further research is needed to clarify the specific role and mechanism of Cav3.2 in CIRI.
In conclusion,our results demonstrate that Cav3.2 KO significantly relieved CIRI-induced oxidative stress,inflammatory response and neuronal apoptosisin vivoandin vitro.Our data suggest that Cav3.2 may be a promising target to combat stroke and CIRI-induced brain injury.
Author contributions:Study design,and manuscript draft:CT,QG,YD;experimental implementation:FD,CH,XL,ZZ,HW,WZ,JW;data analysis:CT,QG,YD,FD,CH,XL,ZZ,HW,WZ,JW.All authors reviewed and approved the final version of the manuscript.
Conflicts of interest:No conflicts of interests are declared by the authors.
Data availability statement:All relevant data are within the paper and its Additional files.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows othersto remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Stephan Kellenberger,Universite de Lausanne,Switzerland.
Additional files:
Additional file 1:Open peer review report 1.
Additional Table 1:Primer sequences for quantitative polymerase chain reaction.
杂志排行

中国神经再生研究(英文版)的其它文章
- Notice of Retraction
- Next-generation regenerative therapy for ischemic stroke using peripheral blood mononuclear cells
- Ruxolitinib improves the inflammatory microenvironment,restores glutamate homeostasis,and promotes functional recovery after spinal cord injury
- OSMR is a potential driver of inflammation in amyotrophic lateral sclerosis
- Optimal transcorneal electrical stimulation parameters for preserving photoreceptors in a mouse model of retinitis pigmentosa
- Neuroprotective effects of G9a inhibition through modulation of peroxisome-proliferator activator receptor gamma-dependent pathways by miR-128