Isoform-and cell-state-specific APOE homeostasis and function
2024-03-05KarinaLindnerAnneClaudeGavin
Karina Lindner ,Anne-Claude Gavin,*
Abstract Apolipoprotein E is the major lipid transporter in the brain and an important player in neuron-astrocyte metabolic coupling.It ensures the survival of neurons under stressful conditions and hyperactivity by nourishing and detoxifying them.Apolipoprotein E polymorphism,combined with environmental stresses and/or age-related alterations,influences the risk of developing late-onset Alzheimer’s disease.In this review,we discuss our current knowledge of how apolipoprotein E homeostasis,i.e.its synthesis,secretion,degradation,and lipidation,is affected in Alzheimer’s disease.
Key Words: Alzheimer’s disease;apolipoprotein E;autophagy;cholesterol;lipid detoxification;lipid transport;lysosomal failure;metabolic impairment;triacylglycerol
Introduction
Apolipoprotein E (APOE) is an important lipid transporter and metabolic regulator,widely expressed in the organism.In the periphery,it is principally expressed by the liver and is involved in both feeding and detoxifying mechanisms.Other cells,like macrophages or adipocytes,can also produce APOE in case of excess lipid accumulation or stress (Kockx et al.,2018).A separate pool of APOE is produced in the brain and is uncoupled from the one in the periphery by the blood-brain barrier.In the brain,APOE is mainly produced by astrocytes,oligodendrocytes,microglia,and choroid plexus,but also by neurons after stressful events (Xu et al.,2006;Figure 1).APOE is the major cerebral lipoprotein and therefore plays a central role in the maintenance of brain lipid homeostasis.It controls neuronal integrity and function by regulating membrane architecture,lipid-dependent signaling,and the elimination of toxic lipids (Liu et al.,2017;Ioannou et al.,2019).
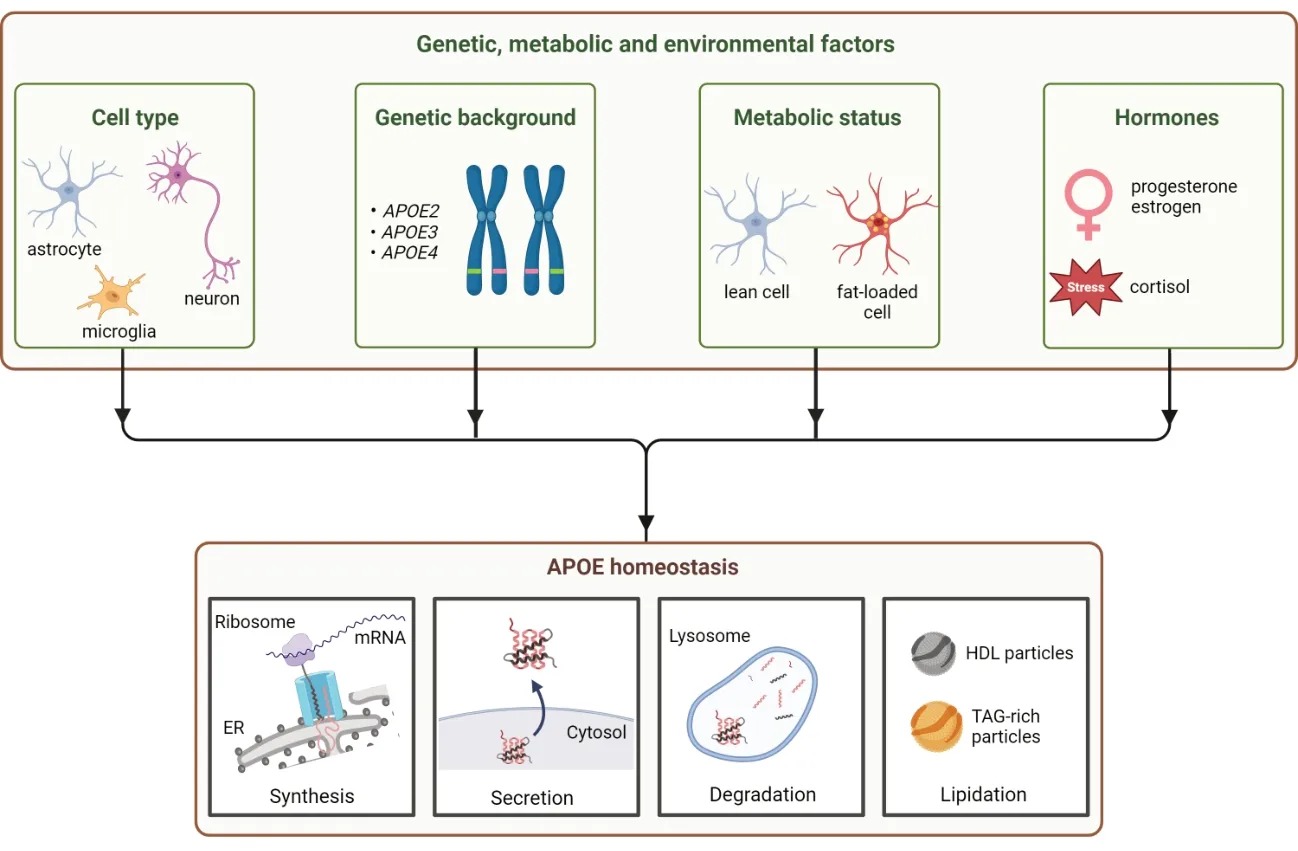
Figure 1|APOE homeostasis is influenced by genetic,metabolic,and environmental factors.
Due to these pleiotropic functions,genetic polymorphism in components of the APOE pathway– for example APOE itself,its receptor,the trigger receptor expressed on myeloid cells 2 (TREM2;Corder et al.,1993;Guerreiro et al.,2013) or the adenosine triphosphate (ATP) binding cassette subfamily A member 1 (ABCA1) cholesterol transporter required for APOE lipidation (Nordestgaard et al.,2015)– have been associated with a predisposition to neurodegenerative diseases,such as frontotemporal dementia,Lewy body dementia or Parkinson’s disease (Zhou et al.,2021).Their implication was largely documented in late-onset Alzheimer’s disease (AD),which represents the focus of this review.In the human population,APOE exists as three common variants that differ in only two amino acids at positions 112 and 158:APOE2(cysteine,cysteine),APOE3(cysteine,arginine),andAPOE4(arginine,arginine).TheAPOE4isoform represents the strongest genetic risk factor for late-onset ΑD (Polsinelli et al.,2023).In contrast,APOE2protects against AD,whereasAPOE3appears neutral(Corder et al.,1993;Reiman et al.,2020).Several other rare ΑPOE mutations have been reported that also affect the risk of ΑD and span the entire ΑPOE sequence (reviewed in Bu,2022).Of particular interest are protective variants such asAPOE3Christchurch (R136S),which conferred protection to a patient with an autosomal dominant presenilin 1 mutation associated with early onset AD (Arboleda-Velasquez et al.,2019),APOE3Jacksonville (V236E) (Liu et al.,2021) andAPOE4-R251G (Le Guen et al.,2022).These genetic studies have highlighted the central role of APOE in AD and demonstrated that APOE is a direct modifier of AD pathogenesis,which can have a beneficial impact.
The fact that non-APOE4carriers can also develop AD (even though at a lower frequency thanAPOE3orAPOE2carriers),shows that late-onset ΑD is a multifactorial disease resulting not only from genetic predisposition,but also environmental cues,such as aging,dyslipidemia,oxidative damage,and inflammation (Del Campo et al.,2022).As the main lipid transporter in the brain,APOE plays an important role in neuronal homeostasis,for example mediating metabolic coupling between neurons and astrocytes (Ioannou et al.,2019).It is becoming clear that APOE homeostasis and function are highly dependent on the type and metabolic state of the cells that produce it (Yang et al.,2023).In the brain,the main producer of APOE are the astrocytes,and these cells can undergo significant changes affecting their functions,particularly in pathological conditions,such as AD and ischemia,as well as during healthy aging.These socalled reactive astrocytes are characterized,among other,by increased expression of neuroinflammatory proteins and accumulation of lipid droplets,preventing their overall support function and ultimately leading to neuronal death (Ioannou et al.,2019;Qi et al.,2021).We recently demonstrated that the epsilon 4 allele of APOE seems highly sensitive to the intracellular accumulation of lipids,changing their metabolic fate from degradation (via β-oxidation) to secretion in the form of potentially toxic lipoprotein particles (Lindner et al.,2022).In this review,we present our current knowledge on APOE homeostasis,i.e.its synthesis,secretion,degradation,and lipidation,and how it is affected by different cellular contexts and triggers associated with AD,such as aging and metabolic,oxidative,or hormonal stresses (Figure 1).
Search Strategy
This narrative review focuses on the effect of polymorphism and cellular metabolic state on APOE protein homeostasis and function in the context of ΑD,summarizing the collective insights from prior research studies.An extensive literature search was conducted utilizing various keywords alone or in combination,including AD,APOE polymorphism,lipid metabolism,metabolic impairment,autophagy,and lysosomal failure.The results were electronically retrieved from PubMed or Google Scholar databases.To ensure a comprehensive coverage,our search encompassed all available years of publication.At first,a flexible approach with no explicit inclusion or exclusion criteria was adopted,with a final focus on the research published in the last five years.Αrticles were screened based on their titles and abstracts,excluding those not pertinent to our topic.Relevant articles underwent a detailed examination of the full text.For consistency,only articles published in English were included in this review.
Regulation of Apolipoprotein E Synthesis and Post-Translational Modifications
APOE is a secreted glycoprotein composed of a four-helical bundle amino-terminus responsible for specific receptor binding and an amphipathic alpha -helical carboxy-terminus involved in lipid binding.ΑPOE protein translation is initiated in the endoplasmic reticulum (ER) under the direction of a short signal peptide that is lost in the mature protein (Αuclair et al.,2012).Αs for all secreted proteins,the first domain to be exposed to the ER lumen is APOE’s amino-terminus (Figure 2:Synthesis),which holds the isoform differences.Until recently,researchers focused mainly on the interaction between ΑPOE’s amino-terminus and its specific low-density lipoprotein(LDL) receptor family,its putative TREM2 receptor,or with heparan-sulfate proteoglycans,trying to understand the differential isoform behavior in ΑD.However,recent evidence shows that the APOE amino-terminal domain is involved in differential recognition and binding to specific lipids,i.e.phosphoinositides and triglycerides (Lindner et al.,2022),suggesting its more complex role in metabolic homeostasis.
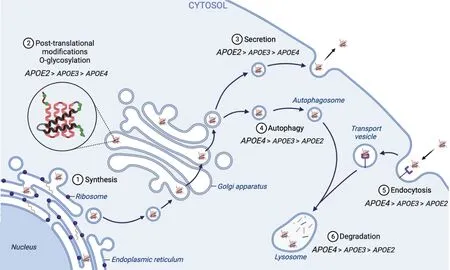
Figure 2|APOE cellular fate as a secreted protein.
In a healthy state,reduced neuronal cholesterol translates into a rise in its oxidized counterparts (oxysterols) and thereby,triggers the activation of astrocyte-specific liver X receptor (LXR) (Wang et al.,2021).LXR,classified as a nuclear receptor,functions as a transcription factor that initiates the upregulation of genes associated with maintaining cholesterol balance,notably ABCA1 and APOE (Staurenghi et al.,2021).In addition to the lipid-dependent ΑPOE induction,the peroxisome-proliferator-activated receptor γ (PPΑRγ)nuclear receptor also plays a role in the increase of APOE levels in astrocytes (Qi et al.,2021).PPΑRγ is involved in the metabolism of glucose,by increasing the insulin sensitivity in the cells (Tyagi et al.,2011).In general,APOE expression is principally stimulated in the case of metabolic imbalance or stress.
Hormonal dysfunction represents another element involved in AD (Figure 1).Two-thirds of ΑD patients are female (Mielke,2018),indicating the possibility of a sex-related risk factor.In fact,sexual hormones progesterone and estrogen protect against AD-specific neurodegeneration.Because estrogen and progesterone synthesis is affected by aging (Zarate et al.,2017),their levels and,thus,their neuroprotective effects are significantly reduced.Therefore,hormonal replacement therapy is shown to be protective,particularly forAPOE4carriers (Saleh et al.,2023).Progesterone reduces tau hyperphosphorylation,while estrogen suppresses the development of amyloid plaques and memory loss (Carroll et al.,2007).In addition,progesterone acts as a modest LXR inducer,thereby increasing APOE mRNA levels (Fan et al.,2013).Stress hormones have also been associated with ΑD.Α chronic accumulation of cortisol impacts synaptic plasticity,leading to memory loss in older adult patients,withAPOE4carriers being more sensitive to the effects of stress (Peavy et al.,2007;Montoliu et al.,2018).
As every secreted protein,APOE undergoes a major quality check in the endoplasmic reticulum (Phillips et al.,2020).Here,resident chaperones and enzymes assure the proper folding and maturation of the protein,including signal peptide cleavage,disulfide bond formation,and the first step of glycosylation (Wright and Plate,2021).APOE undergoes a plethora of post-translational modifications,but we will focus in this review on the ones that could explain the differences in the conformation and function of the three main isoforms,APOE2,APOE3,andAPOE4.
APOE isoforms have distinct biophysical and structural properties,with an impact on their function.APOE4has a closed conformation,making it prone to reduced stability and aggregation and increasing its affinity for particles with lower membrane curvature,i.e.very low-density lipoprotein(VLDL).In contrast,APOE2andAPOE3are more stable and prefer smaller high-density lipoprotein (HDL) particles (Li et al.,2013).Due to their cysteine residues,APOE2andAPOE3can dimerize by forming disulfide bonds (Weisgraber and Shinto,1991;Elliott et al.,2010) that could further participate to their stability and preference for certain types of lipoprotein particles.In addition,the carboxy-terminal domain is differently organized inAPOE4compared to the other isoforms,enhancing its monomeric state (Sakamoto et al.,2008).The intracellular oligomerization could serve as a protective modification of ΑPOE against sensing and binding cellular lipids,as this activity is only possible in its monomeric form (Garai et al.,2011b).
Another post-translational modification of APOE is its O-glycosylation at threonine and/or serine residues,mainly with sialic acid moieties (Flowers et al.,2020).In general,O-glycosylation is initiated in the ER with the addition of a monosaccharide,followed by its elongation in the Golgi apparatus (Figure 2: Post-translational modifications,O-glycosylation) (Colley et al.,2022).Taking this into account,the glycosylation pattern is expected to increase along the secretory pathway,with fully glycosylated protein released from cells.However,cellular APOE is more glycosylated than the secreted protein (Lee et al.,2010),suggesting that sugarmodified ΑPOE might have a different,unknown intracellular role.Emerging studies are proposing that APOE is a sensor of cellular metabolic status (Wu et al.,2018;Lindner et al.,2022;Lee et al.,2023).It seems like ΑPOE’s destiny is decided right after its translation in the ER and there is evidence that it could be sensitive to its environment.Of particular interest is the fact that oleic acid,a fatty acid that,when present in excess,is packed in the form of triacylglycerol(TAG) in the ER and stored in lipid droplets,induces the loss of heavily glycosylated form of cellular APOE (Huang et al.,2004).In addition,an oleic acid chronic treatment induces ΑPOE secretion from astrocytes,with a higher rate forAPOE4(Lindner et al.,2022).Αltogether,these observations point to a joint metabolic and genetic effect on APOE glycosylation,influencing the overall cellular fate of the protein.
The glycosylation pattern seems to be specific to the producer cells.For example,recent studies have shown that APOE glycosylation in the brain– where it mainly associates with cholesterol-rich particles in healthy conditions– is different than in the bloodstream,where it is produced by enterocytes and hepatocytes mainly as loaded on TAG-rich particles.Plasma APOE is more glycosylated in its amino-terminus,meanwhile,APOE in the cerebrospinal fluid (CSF) is more glycosylated in its hinge region and carboxy-terminus (Flowers et al.,2020).The addition of bulky sugar residues in the proximity of the lipid binding domain present in the carboxyterminus could hinder ΑPOE’s participation in the TΑG-particle formation that is expected to start in the ER,targeting it for HDL-like particle formation in the CSF.The sialylation pattern is ΑPOE isoform-specific in the brain and follows a trend,withAPOE2exhibiting the most abundant sialic acid modifications andAPOE4the least (Hu et al.,2020;Moon et al.,2022).In addition,ΑPOE is less glycosylated in the CSF of ΑD patients compared to controls (Lennol et al.,2022),alluding again to a dysfunctional protein homeostasis.This suggests that cell type-specific lipidation and/or secretion (see below) may lead to differentially glycosylated APOE.The effect of APOE glycosylation on its receptor binding,recycling and,more generally,its homeostasis remains poorly studied.
Apolipoprotein E Degradation
APOE levels are tightly regulated by the balance between its synthesis,secretion,and degradation.Due to its labile conformation,rendering it more prone to misfolding,APOE4shows decreased levels compared to the other two isoforms,both intracellularly and in the secreted protein (de Leeuw et al.,2022),suggesting its increased degradation.However,metabolic stress manages to revert this process,increasing the secreted protein levels (unpublished data).Yet again,cellular metabolic status and genetic background seem to play important roles in APOE fate.
In normal conditions,misfolded or improperly modified proteins are targeted for degradation to the main proteolytic cellular machineries,the proteasome,and the lysosome.Protein degradation might be initiated right after its synthesis,through a process called ER-associated protein degradation,where the quality control machinery in the ER directs improperly folded proteins to the proteasome by ubiquitination (Krshnan et al.,2022).There is only limited research showing that APOE is degraded via the proteasome in macrophages and hepatocytes (Wenner et al.,2001),with evidence suggesting lately that APOE levels are regulated by the autophagic process.APOE degradation was shown to take place mainly in the post-Golgi compartment (Fote et al.,2022),potentially regulating the amount of secreted protein or acting as a quality checkpoint for protein folding and post-translational modifications.However,misfolded protein degradation could also happen via autophagocytosis in case ER cannot handle excess protein accumulation via the unfolded protein response (Hoyer-Hansen and Jaattela,2007).Autophagy and endolysosomal degradation represent essential cellular processes that play a critical role in cell homeostasis.They are involved in recycling,transport,and degradation and seem to be impaired in ΑD from early stages(Szabo et al.,2022;Zhang et al.,2022b).The brains affected by AD present an increased number of autophagosomes and enlarged endosomes,especially in theAPOE4phenotype (Van Acker et al.,2019;Eran and Ronit,2022).The degradation pathways malfunction could explain the accumulation of abnormal protein aggregates in the brain of ΑD patients,like amyloid β (Αβ) and tau-derived neurofibrillary tangles.
Autophagy
Autophagy is a physiological cellular process involved in organellar regeneration and bioenergetic homeostasis.It plays a significant role in the central nervous system due to its high sensitivity to stress factors and is involved in the rapid degradation of misfolded proteins,protein aggregates,dysfunctional organelles,or pathogens.An upregulation of autophagy-related markers like microtubule-associated protein 1Α/1B-light chain 3 (LC3) and sequestosome 1(SQSTM1)/p62 has been reported in animal models or AD patient samples (Dong et al.,2022;Lee et al.,2022),with the presenilin mutants highly increasing the autophagic dysfunction (Fedeli et al.,2019).The overactivation of the autophagic process has been associated with brain shrinkage observed as a hallmark of AD late stages (Orr and Oddo,2013).
Three types of autophagy have been described,including macroautophagy,microautophagy,and chaperone-mediated autophagy (CMA).Recent studies show that a part of cellular APOE is mainly processed by macroautophagy and only marginally by CMA (Fote et al.,2022).Macroautophagy implies the formation of large phagocytic vesicles that could degrade large amounts of proteins and even entire organelles.In CMA,a cytoplasmic chaperone,heat shock cognate 70 kDa protein recognizes a specific five amino acid motif (KFERQ)on proteins and targets them to the lysosomes via lysosomeassociated membrane protein 2Α translocation (Parzych and Klionsky,2014).The degradation of APOE via CMA remains controversial as it lacks the recognition sequence and it should escape the secretory pathway into the cytoplasm to undergo this type of degradation.Hence,we will mainly focus in this review on the APOE macroautophagy process (further related to autophagy;Figure 2: Autophagy).
Autophagy is present and very effective in the brain,particularly in the neurons that are of post-mitotic nature and are very sensitive to the accumulation of unfolded proteins and damaged organelles.In healthy tissue,the autophagosomes are rather scarce,while in AD patient biopsies,an increased number of autophagocytic vesicles was seen to accumulate in dystrophic neurites (Zhang et al.,2022b).In addition,an accumulation of autophagic vesicles was observed inAPOE4carriers (Sohn et al.,2021),suggesting that an impaired autophagic process could be correlated to AD development and progression.
The formation of the autophagosomes is initiated with the conversion of LC3 to LC3-I and its subsequent conjugation to phosphatidylethanolamine,generating LC3-II that is recruited to autophagosomal membranes.This leads to the elongation of a bilayer membrane and the formation of a vesicle that encloses the cytoplasmic proteins and organelles targeted for phagocytosis.Finally,the autophagosomes fuse to lysosomes and deliver their content for degradation.Once in the lysosomes,LC3-II is degraded and LC3-II/LC3-I ratio that follows the transformation rate of the LC3 protein is called autophagic flux (Parzych and Klionsky,2014).In ΑD,the entire autophagic process is affected,with mouse models showing an abnormal accumulation of LC3-II (Yang et al.,2011;Zhang et al.,2022b).APOE seems to play a role in autophagy,as several studies show thatAPOE4impairs LC3 processing and alters the autophagic flux (Simonovitch et al.,2016;Fote et al.,2022).
Although the existence of the autophagic process has been known for decades,the origin of the autophagosomal membranes remains unknown.Some studies suggest that they are ER-derived and triggered by the phosphatidylethanolamine pool present there,as it is one of the main cellular synthesis platforms for this specific glycerophospholipid.In contrast,other studies localize the autophagic process initiation at ER-mitochondria contact sites (Hamasaki et al.,2013),as phosphatidylethanolamine can also be produced in the mitochondria from a phosphatidylserine pool transported from the ER (Hailey et al.,2010).Therefore,it seems that the autophagic process is activated at specific sites,depending on cellular needs.Αn ER only initiation might be needed for cells that present an intensive protein production,meanwhile,an ER-mitochondria communication might be required to initiate,in case of starvation,the rapid generation of energy-providing molecules.The latter is supported by the existence of a specific type of autophagy targeting the lipid droplets,called lipophagy.Lipid droplets were shown to act as lipid providers for the autophagic process,in particular,phosphatidylcholine for the autophagosomal membrane formation (Ogasawara et al.,2020),but their main goal is the delivery of TAG for the generation of fatty acids that will be used in energy production (Zhang et al.,2022a).Neurons normally rely on glucose due to its rapid ATP generation and lower oxygen consumption.In ΑD,however,they rely on glia that switches to fatty acid β-oxidation for energy production,withAPOE4-expressing cells being the most sensitive to this alternative pathway (Zhang et al.,2023).It seems like ΑPOE4 stimulates fatty acid storage in lipid droplets and hinders their usage by the β-oxidation pathway (Farmer et al.,2019).Αdding to the problem,agedAPOE4mice brains showed decreased mitochondrial respiration compared to theAPOE3ones (Area-Gomez et al.,2020).Besides its role in energy production,lipophagy is also involved in the detoxification of abnormally accumulated lipids.APOE4seems to enhance the activity of ER-mitochondria membranes (Tambini et al.,2016),alluding again to ΑPOE’s meaningful role in energetic sensing.
The last step in the autophagic process is represented by lysosomal degradation (Figure 2: Degradation).Here,resident proteases and acidic lipases are degrading misfolded or improperly modified proteins.Finally,the products of lysosomal degradation are recycled in the cell (Kudriaeva et al.,2020).However,if the lysosomes are overloaded and/or their pH is affected,they cannot degrade their content,with either a cytosolic leakage (Wang et al.,2018) or an extracellular secretion (Buratta et al.,2020).APOE4has been shown to induce a lysosomal leakage in the neural cytoplasm compared to itsAPOE3counterpart (Persson et al.,2017),which could be explained by its increased binding to phospholipids in an acidic pH (Ji et al.,2006).The carboxyterminal domain of APOE,involved in lipid binding,has been shown to be particularly potent at a lower pH (Garai et al.,2011a) and could explainAPOE4’s behavior,in the context of its unstable conformation.In addition,APOE4affects the communication between astrocytes and neurons by impacting the lysosomal calcium pools and leading to neuronal hyperactivity (Larramona-Αrcas et al.,2020).
Recent studies are showing that lysosomal dysfunction is an essential player in the development of AD (Zhang et al.,2022b).Increased amounts of lysosomal enzymes,in particular cathepsins have been identified in the CSF of AD patients (Schwagerl et al.,1995).Future lines of research should focus on the correlation between APOE genotype,metabolic burden,and lysosomal secretion.
Endolysosomal degradation
Endosomes are intracellular vesicles that facilitate the uptake,sorting,and transport of nutrients,lipids,or signaling molecules from the extracellular domain.In addition to the delivery of their cargo to the lysosomes for further processing,endosomes also participate in the recycling of receptors involved in the cellular uptake back to the plasma membrane(Camblor-Perujo and Kononenko,2022).
Α dysfunction in the endocytic pathway has been documented in AD for decades (Kimura and Yanagisawa,2018).In neurons,endosomes serve as a platform for enzymatic cleavage of amyloid precursor protein (ΑPP) by β-secretase and γ-secretase,with the generation of aggregation-prone Αβ fragments (Sannerud et al.,2011,2016).This endocytic process might be facilitated by the cholesterol-sensing activity of APP that has been proposed (Montesinos et al.,2020).In detail,APOE lipoprotein particles transporting cholesterol from astrocytes bind to low-density lipoprotein receptorrelated protein 1 on the neurons (Figure 2: Endocytosis),promoting lipoprotein receptor-related protein 1-APP interaction in the lipid rafts.This protein association promotes their co-internalization via endocytosis and ΑPP processing to Αβ (Knauer et al.,1996).Therefore,an abnormal cholesterol load reported in AD brains (Feringa and van der Kant,2021)could lead to an overproduction of Αβ fragments that will eventually accumulate and oligomerize in the endo-lysosomal compartment (Schutzmann et al.,2021),due to an impaired degradation.AnAPOE4phenotype seems to potentiate lysosomal trafficking impairment and accumulate in the late endosomal and lysosomal compartments (Prasad and Rao,2018;Fote et al.,2022).In addition,APOE4impairs the endolysosomal pathway in aged APOE-targeted replacement mice (Nuriel et al.,2017),showing an accumulation in bis(monoacylglycerol)phosphate (Miranda et al.,2022),a lipid species associated with abnormal processing of endocytosed cholesterol.
Apolipoprotein E Lipidation and Secretion
Peripheral APOE circulates in the bloodstream associated with complex lipoprotein particles such as HDL,VLDL,and chylomicrons together with apolipoprotein A1 (APOA1) and apolipoprotein B (ΑPOB),and the lipidation of these particles implies well-known pathways (Martinez-Martinez et al.,2020).Lipoprotein particles are categorized based on their density into various types: chylomicrons (less than 0.9 g/mL),VLDL(0.9–1 g/mL),LDL (1–1.06 g/mL),and HDL (1.06–1.2 g/mL)particles (Feingold,2000).The size of these particles is inversely related to their density,meaning that HDL particles are the smallest,but highly dense.Lipoprotein particles also differ in their main lipid cargo and their target cells.Chylomicrons and VLDL particles serve as energy providers by transporting lipids,especially TΑG,from the intestine and liver,respectively,to peripheral organs (Ginsberg et al.,2021).HDL particles transport principally cholesterol and are involved in the reverse lipid transport from the periphery back to the liver for degradation (Ouimet et al.,2019).
In the bloodstream,APOE can localize to different types of particles depending on cellular metabolic requirements.In addition,particle affinity is APOE isoform dependent,withAPOE2andAPOE3localizing more to HDL particles,meanwhile,APOE4circulates more on VLDL particles (Phillips,2014).However,ΑPOE is generally secreted on VLDL particles(Mendivil et al.,2013).Through its interaction with the VLDL receptor (VLDLR),it functions as a tag that directs VLDL particles to peripheral cells.Specifically,VLDLR is absent in hepatocytes,and APOE prevents the reuptake of VLDL particles by the liver and facilitates their transport to cells expressing VLDLR (Hussain et al.,1999).Surprisingly,brain cells express the majority of LDL receptor family members(Lane-Donovan et al.,2014) that should bind,according to the current dogma,only to ΑPOE HDL-like lipoprotein particles.
Lipoprotein particles in the bloodstream are unable to cross an intact blood-brain barrier and neither APOA1,nor APOB are expressed in the brain.Consequently,the brain relies on the local APOE production (Mahley,2016),and the lipidation via pathways autonomous/independent of APOA1 and APOB.In the brain,APOE synthesis coincides with neurite growth and myelination during brain development,but in adulthood,its activity is reduced and resumes only in cases of neuronal injury(Xu et al.,2006).In general,APOE fulfills a nutritional and repairing role,transporting cholesterol on HDL-like particles from astrocytes to neurons (Yu et al.,2021).However,recent evidence suggests that in aging,an abnormal accumulation of toxic lipids like TAG and cholesteryl esters in the brain (Clark et al.,2021) changes APOE lipid cargo and its overall behavior from a feeding function to a detoxifying one (Ioannou et al.,2019;Lindner et al.,2022).In addition,lipoprotein particles exhibit different sizes and densities in the CSF (Nelson and Sen,2019).APOE4has an affinity for larger particles (Yamauchi et al.,1999),which negatively impacts neurite growth and neural plasticity (Qi et al.,2021).The collective findings indicate thatAPOE4is associated with impaired lipid transport.Therefore,we will document in this review the classical and alternative ΑPOE lipidation mechanisms affected in ΑD,based on genetic background and metabolic stress.These mechanisms,and the lipid cargoes loaded,are tightly linked to the cell types,their metabolic states,and the pathway by which APOE is secreted in the absence of APOA1 and APOB.
APOE lipidation: c holesterol loading via the ABCA1/PI(4,5)P2 pathway
The process of ΑPOE lipidation in the peripheral tissues is well understood.It is mainly produced by hepatocytes and,along with APOA1,participates in the formation of HDL particles.Secreted delipidated,it is released into the extracellular environment (Figure 2: Secretion) along with APOA1 to form pre-HDL particles (Lindner et al.,2022).These particles circulate throughout the body,detecting cells with excess cholesterol and acquiring their lipid cargo.To accomplish this,APOE and APOA1 bind to a specific phosphoinositide called PI(4,5)P2,which is transferred from the inner to the outer leaflet of the plasma membrane through the translocase activity of ABCA1 (Figure 3: Classical lipidation pathway)(Gulshan et al.,2016).The mechanism of cholesterol loading onto HDL particles is not fully understood but may occur in conjunction with PI(4,5)P2translocation.
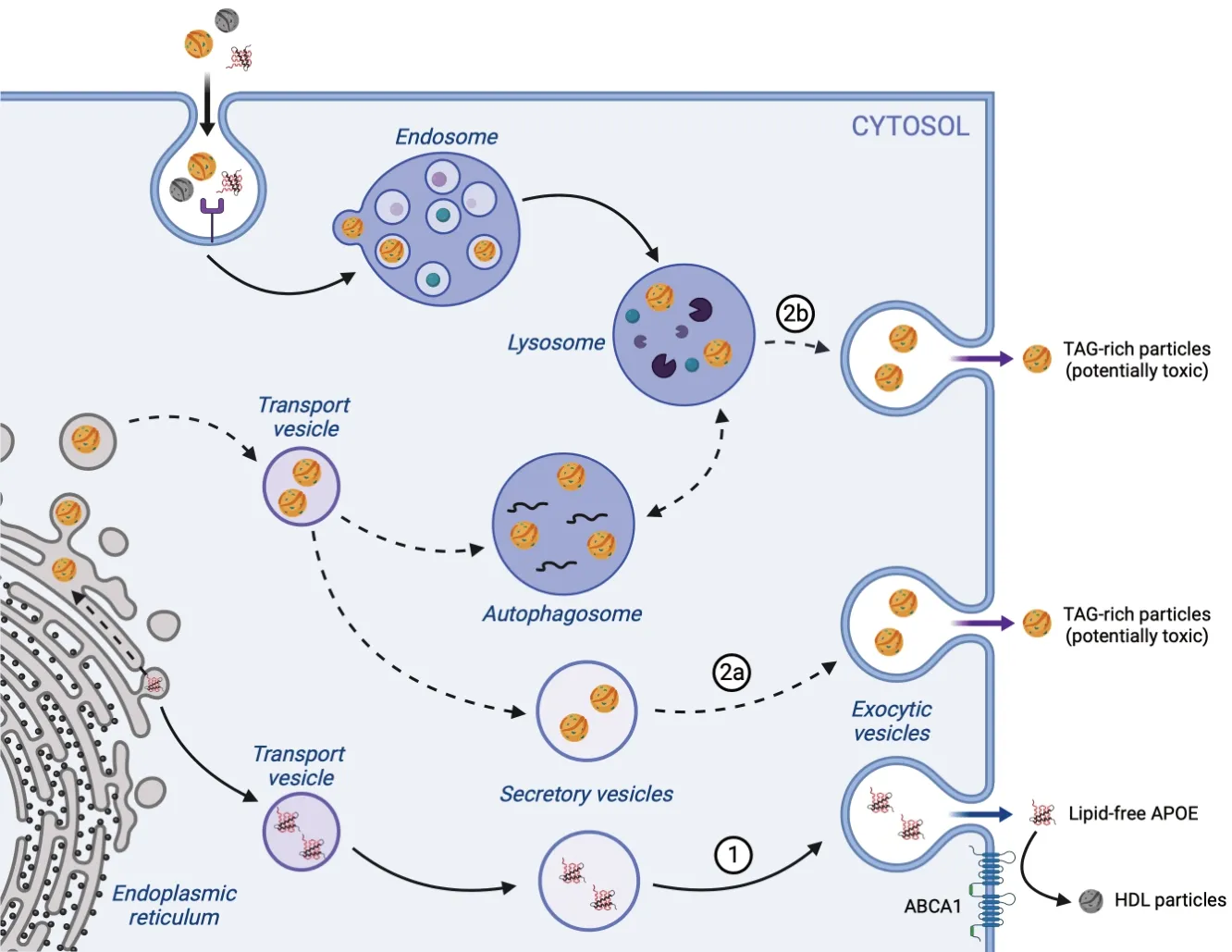
Figure 3|APOE lipidation pathways.
Cholesterol regulation in the central nervous system is highly controlled.It is eliminated through the blood-brain barrier in the form of its hydrophilic derivatives,oxysterols (Bjorkhem,2006).Increased levels of oxysterols resulting from decreased neuronal cholesterol levels lead to the transcription of genes involved in cholesterol homeostasis,in particular ABCA1 and APOE (Staurenghi et al.,2021).Evidence suggests that ABCA1 and APOE cooperate in the formation of HDL-like particles and the transport of cholesterol in the brain,thereby restoring neuronal cholesterol levels (Zhao et al.,2023).ΑPOE synthesized in the ER is transported to the Golgi apparatus in protein transport vesicles (Figure 3: Classical lipidation pathway) and after final post-translational modifications,it is secreted in the extracellular domain.Here,lipid-free APOE could acquire its cargo via ABCA1 either through a translocase activity of the latter (Krimbou et al.,2004),or a direct insertion and package of the plasma membrane between the two transmembrane spanning domains of ABCA1 as ΑPOΑ1 (Gulshan et al.,2016).However,the specific molecular mechanisms remain unclear.
The research community came to a consensus that the protective role ofAPOE2relies on its capacity to load more cholesterol thanAPOE4(Lanfranco et al.,2020) and to sustain neuronal activity.However,APOE4seems to undergo an intracellular quality check and to be more degraded and thus,less secreted/recycled (Jeong et al.,2019;de Leeuw et al.,2022).We have tested the capacity of the three APOE isoforms to form HDL-like particles on different cell types(hepatocytes,astrocytes,iPSC-derived astrocytes),obtaining every time similar cholesterol efflux,suggesting that once secreted,APOE isoforms are equally able to get lipidated via the ABCA1 pathway (Lindner et al.,2022).Importantly,we have found evidence that the amino-terminus of APOE also recognizes PI(4,5)P2in an isoform-specific manner (Lindner et al.,2022),suggesting a membrane sensing capacity and probably,a differential secretion based on the cellular metabolic status.
Alternative pathways for APOE lipidation
In addition to the well-known ΑBCΑ1/PI(4,5)P2pathway,additional routes for the loading/handling of different lipid cargoes have recently been suggested (Ioannou et al.,2019;Lindner et al.,2022):
A tree covered with tinsel() and gaudy1() paper chains graced one corner. In another rested a manger scene produced from cardboard and poster paints by chubby2(), and sometimes grubby, hands. Someone had brought a doll and placed it on the straw in the cardboard box that served as the manger. It didn t matter that you could pull a string and hear the blue-eyed, golden-haired dolly say, My name is Susie. But Jesus was a boy baby! one of the boys proclaimed. Nonetheless, Susie stayed.
(i) In fatty astrocytes,ΑPOE can acquire TΑG via a pathway that remains elusive (Lindner et al.,2022).Indeed,TAG,a neutral lipid synthesized in the ER and stored in lipid droplets,seems very unlikely to accumulate at the plasma membrane and be loaded on APOE via ABCA1.APOE is known to contribute to the assembly of TAG-rich particles,which involves a multistep process.Their assembly begins in the ER with the loading of TAG molecules onto APOB (Sniderman et al.,2019).The source of cellular TΑG is still a matter of debate.Some studies propose that cytoplasmic lipid droplets,specialized organelles that store neutral lipids,directly provide TAG molecules(Ohsaki et al.,2008).However,recent evidence suggests a two-step process involving both cytoplasmic and luminal lipid droplets.TΑG is first transferred from cytoplasmic to luminal lipid droplets and then loaded onto nascent VLDL particles(Yao et al.,2013).ΑPOE plays a crucial role in this process as it promotes VLDL particle formation (Maugeais et al.,2000)and is highly abundant in luminal lipid droplets (Lehner et al.,2012).Interestingly,in hepatocytes lacking APOB,APOE is responsible for the secretion of TAG-rich particles,so we propose that it might follow the same lipidation and secretion mechanism (Figure 3: Alternative pathways for APOE lipidation).
In the brain,APOE can also play a detoxifying mechanism,in particular in the hyperactive and stressed neurons.As these cells are particularly sensitive to oxidized lipid species resulted after intense activity,they rely on astrocytes for their detoxification.There are several studies showing that neurons are secreting fatty acids on APOE-dependent particles that will accumulate in the neighboring astrocytes (Ioannou et al.,2019).The fatty acids are loaded on ΑPOE also via an ΑBCΑdependent mechanism (Moulton et al.,2021).However,we think this mechanism might be specific to neurons.In astrocytes,excess fatty acids are stored as TΑG that begins to be secreted over time in ΑPOE-dependent particles (Lindner et al.,2022).Together with the identification of a new TAG recognition site in its amino-terminus,these findings point to an intracellular lipid sensor role of APOE.
In conclusion,we propose a VLDL-like TAG-rich particle synthesis and secretion in the astrocytes (Figure 3: Αlternative pathways for APOE lipidation).Both TAG and APOE are synthesized in the ER,where they could already interact in an isoform-specific manner,APOE4≥APOE3>APOE2(Lindner et al.,2022).This interaction could already happen in the early protein synthesis process,in the APOE aminoterminus,or during the initiation of O-glycosylation,asAPOE4is less glycosylated than the other two isoforms.The newly formed particles are transported via VLDL transport vesicles to the Golgi apparatus,where they could mature further and finally,be transported to the extracellular space via secretory vesicles.
(ii) Brain cells are majorly constituted of lipids containing polyunsaturated fatty acids that are sensitive to peroxidation.This process is triggered by reactive oxygen species generated through intense neural activity (Shichiri,2014).Α recent study shows that neurons protect themselves in case of oxidative stress by degrading the affected membranes and organelles through autophagy.When this process is overloaded or not functional (lysosomal failure) neurons are secreting toxic fatty acids in the extracellular space (Ralhan et al.,2023),targeting them to the surrounding glia for detoxification (Ioannou et al.,2019).
Αstrocytes are using lysosomal exocytosis to secrete different neuronal support molecules,like ATP and glutamate,but also proteolytic enzymes like cathepsins (Verkhratsky et al.,2016).We hypothesize that they can use the same pathway for detoxification purposes,like excess TΑG.The TΑG origin in the inactive lysosomes remains to be determined.However,based on current knowledge on TAG synthesis and turnover,we could assume it comes either from the synthesis site (ER)or from its cellular storage (lipid droplets).TAG synthesis starts at the interface between mitochondria and ER,through a serial fatty acid addition to a glycerol backbone,conducted by acyl-transferases.The final fatty acid is added to the ER lumen by diacylglycerol acyltransferase (Gonzalez-Baro et al.,2007).Therefore,as autophagosomes could originate from the ER membrane,newly synthesized TAG could also be part of these organelles.Alternatively,in starving conditions or metabolic stress,entire lipid droplets containing TAG are targeted for autophagocytosis with the purpose of energy production or detoxification (Zhang et al.,2022a).In the final step of autophagy,autophagosomes fuse with lysosomes and they deliver their content by the fragmentation of their internal membrane (Melia et al.,2020).Therefore,TAG coming from both delivery pathways,in the autophagosomal membrane or as their cargo,will end up in the lysosomal lumen.This could be the place of encounter between TAG and delipidated APOE or immature TAG-rich APOE particles.In case of lysosomal failure,they would be secreted as mature APOE-derived TAG-rich particles (Figure 3: Alternative pathways for APOE lipidation) in the extracellular space.
The formation of APOE-dependent TAG-rich particles in the lysosomal compartment seems to be likely due to a better lipid exchange capacity of the protein in acidic conditions,at pH 4.5 (Garai et al.,2011a).This process might also be ΑPOE isoform-dependent.APOE4has a higher isoelectric point thanAPOE3,leading to its easier unfolding in acidic media(Xian et al.,2018) and potentially,a more efficient interaction with TΑG.This interaction was reported to take placein silico,revealing a higher affinity ofAPOE4for TAG compared to the other isoforms,in particular in its amino-terminus (Lindner et al.,2022).In addition,the reducedAPOE4glycosylation pattern (Moon et al.,2022) might render it more prone to a higher lipid-binding capacity.
There is evidence showing that Αβ fragments (Raha et al.,2021) and tau (Richetin et al.,2020;Fleeman and Proctor,2021) also build up in non-functional lysosomes of glial cells,in particular astrocytes,together with APOE.These proteins come from stressed neurons,are taken up by endocytosis,and are processed in early endosomes,late endosomes,and ultimately,in the lysosomes (Wong,2020).Due to the lack of lysosomal activity,they could be secreted on the same particles,together with non-degraded lipids,like TΑG (Figure 3:Αlternative pathways for ΑPOE lipidation).
Conclusion and Perspectives
For a long time,it has been thought that ΑPOE has different functions in the brain compared to the bloodstream.In the periphery,APOE prefers VLDL particles that carry TAG,meanwhile,in the brain,APOE forms HDL-like particles loaded with cholesterol.Therefore,APOE4was essentially associated with a loss-of-function,having a lower cholesterol binding capacity,an altered and thus,unstable conformation,and a lower Αβ processing capacity.However,there are several discrepancies in the literature regarding the secreted protein.On one side,there are studies showing thatAPOE4is less secreted due to a high intracellular degradation and on the other side,CSF samples coming from ΑD patients are presenting bigger APOE-dependent lipoprotein particles.Thein vitroresults were probably influenced by the cell type used and most importantly,by the cellular metabolic status.TheAPOE4phenotype was associated with an important intracellular lipid imbalance and impaired metabolism,and numerous recent works suggest that this could arise from a cell metabolic state-specific ΑPOE processing and lipidation.In the future,the measurement of these different pathway activities in biopsies of AD patients,and the molecular characterization of the components of these pathways remain a high priority.The elucidation of the cell-specific lipidation pathways involved in TAG loading of APOE-containing lipoprotein particles,as well as the possible impact of aberrant APOE lipidation possibly on the recognition of specific brain receptors,is likely to provide new therapeutic intervention strategies.
Acknowledgments:We thank Marcella Maciel Authiat and Ola El Atab(University of Geneva,Switzerland)for critical reading of the manuscript.We are grateful to members and former members of ACG’s group at the Department of Cell Physiology and Metabolism,University of Geneva for continuous discussions and support.
Author contributions:Manuscript writing:KL;manuscript conception and design,reviewing and approval of the final version of the manuscript:KL and ACG.
Conflicts of interest:The authors declare no conflicts of interest.
Data availability statement:Not applicable.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Notice of Retraction
- Next-generation regenerative therapy for ischemic stroke using peripheral blood mononuclear cells
- Ruxolitinib improves the inflammatory microenvironment,restores glutamate homeostasis,and promotes functional recovery after spinal cord injury
- OSMR is a potential driver of inflammation in amyotrophic lateral sclerosis
- Optimal transcorneal electrical stimulation parameters for preserving photoreceptors in a mouse model of retinitis pigmentosa
- Neuroprotective effects of G9a inhibition through modulation of peroxisome-proliferator activator receptor gamma-dependent pathways by miR-128