The physiological role of the unfolded protein response in the nervous system
2024-03-05ShuangchanWuWenshengLin
Shuangchan Wu ,Wensheng Lin,*
Abstract The unfolded protein response (UPR) is a cellular stress response pathway activated when the endoplasmic reticulum,a crucial organelle for protein folding and modification,encounters an accumulation of unfolded or misfolded proteins.The UPR aims to restore endoplasmic reticulum homeostasis by enhancing protein folding capacity,reducing protein biosynthesis,and promoting protein degradation.It also plays a pivotal role in coordinating signaling cascades to determine cell fate and function in response to endoplasmic reticulum stress.Recent research has highlighted the significance of the UPR not only in maintaining endoplasmic reticulum homeostasis but also in influencing various physiological processes in the nervous system.Here,we provide an overview of recent findings that underscore the UPR’s involvement in preserving the function and viability of neuronal and myelinating cells under physiological conditions,and highlight the critical role of the UPR in brain development,memory storage,retinal cone development,myelination,and maintenance of myelin thickness.
Key Words: myelin;neuron;oligodendrocyte;Schwann cell;unfolded protein response
Introduction
One-third of the proteome is directed to the endoplasmic reticulum (ER) in eukaryotic cells,where modification and folding of membrane and secretory proteins take place.Protein modification and folding inside the ER lumen is highly sensitive to the imbalances between the rate of mRNΑ translation and the efficiency of protein folding.Such imbalances can result in failure of protein modification and folding (Costa-Mattioli and Walter,2020;Lin and Stone,2020;Marciniak et al.,2022).Indeed,a significant percentage of protein modification and folding in the ER will be failed.Many proteins,particularly those with multiple transmembrane regions,have a propensity to spontaneously unfold,misfold,or aggregate during maturation in the ER.Only a small fraction of these proteins meet the requirements of the ER protein quality control mechanism (Balch et al.,2008;Martinez et al.,2018).The aggregates of unfolded or misfolded proteins within the ER lead to toxic consequences for the organism,which has been termed to as ER stress (Lin and Popko,2009;Marciniak et al.,2022;Wiseman et al.,2022).ER stress is further managed through the activation of the unfolded protein response (UPR),as depicted inFigure 1.Its primary goal is to reestablish ER homeostasis and to enable cellular adaptation to ER stress by promoting protein folding,reducing protein translation,and enhancing protein degradation (Walter and Ron,2011;Lin and Stone,2020;Wiseman et al.,2022).Nevertheless,if these adaptive measures fail to reestablish ER homeostasis,persistent activation of the UPR can trigger apoptosis programs to eliminate the stressed cells (Marciniak and Ron,2006;Lin and Stone,2020;Wiseman et al.,2022).
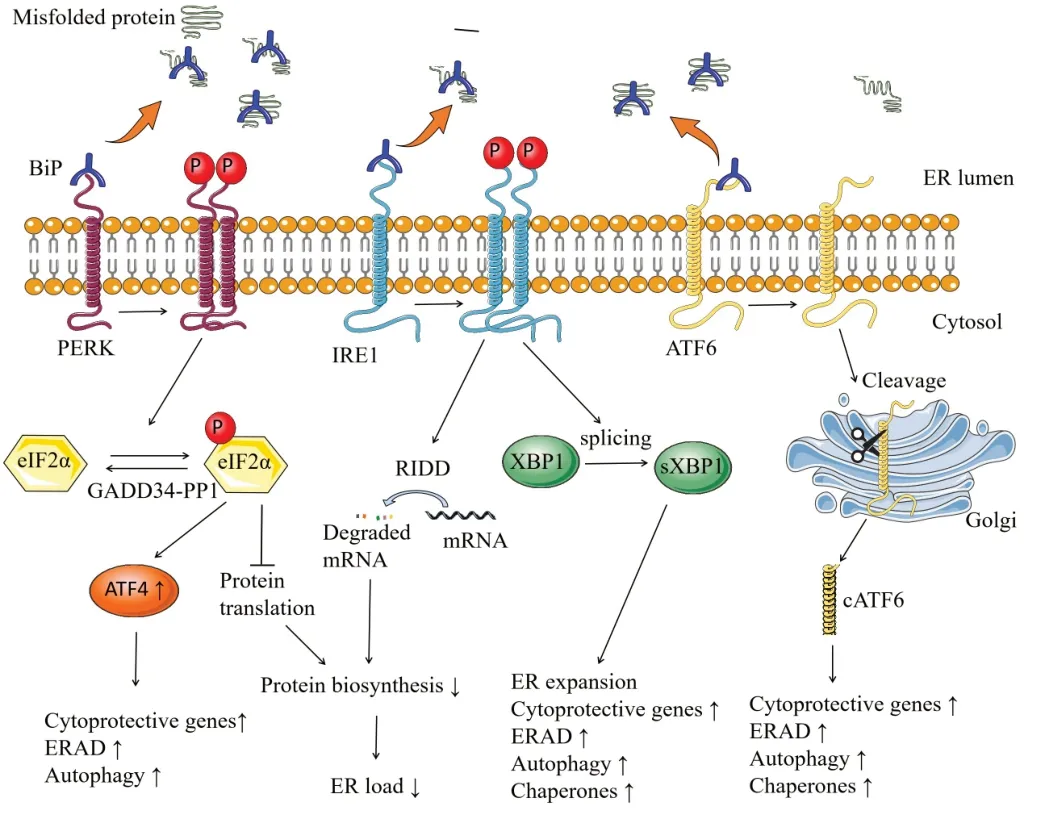
Figure 1|Schematic diagram of the UPR.
To maintain ER homeostasis,the UPR is initiated by parallel signaling networks,including three major ER stress sensors:pancreatic ER kinase (PERK),inositol requiring enzyme 1 (IRE1),and activating transcription factor 6 (ATF6) (Marciniak and Ron,2006;Lin and Stone,2020;Marciniak et al.,2022).Under physiological conditions,binding immunoglobulin protein (BiP,also known as GrP78) binds to the luminal domain of PERK,IRE1 and ATF6,maintaining the transmembrane proteins in an inactive conformation (Martinez et al.,2018;Lin and Stone,2020;Wiseman et al.,2022).Upon ER stress,BiP is recruited by unfolded or misfolded proteins to avoid them aggregation,resulting in the releasing the UPR transducers to allow their activation (Lin and Stone,2020;Marciniak et al.,2022).
When ER stress initiates,the PERK pathway is engaged as the most rapidly activated signaling.The dissociation of BiP results in the activation of PERK through oligomerization and autophosphorylation.Phosphorylation of PERK inhibits global protein translation by phosphorylating α subunit of eukaryotic translation initiation factor 2 (eIF2α),resulting in a reduced ER load (Stone and Lin,2015;Costa-Mattioli and Walter,2020;Wiseman et al.,2022).The eukaryotic translation initiation factor 2B (eIF2B) functions as a guanine nucleotide exchange factor,aiding in the release of GDP from its substrate,eIF2.This action consequently triggers the formation of an active eIF2-GTP complex.Subsequently,the eIF2-GTP complex associates with aminoacylated initiator methionyl tRNA,leading to the establishment of a ternary complex crucial for the initiation of protein translation (Pavittand Proud,2009;Lin and Stone,2020).Phosphorylated eIF2α (p-eIF2α) suppresses the activity of eIF2B by forming a nonproductive p-eIF2α–eIF2B complex (Pavitt and Proud,2009;Hanson et al.,2022).Conversely,p-eIF2α promotes the translation of specific mRNΑs that harbor upstream open reading frame sequences within their 5′ untranslated regions(Lu et al.,2004a;Taylor and Hetz,2020),including activating transcription factor 4 (ΑTF4).ΑTF4 induces the expression of certain cytoprotective genes,autophagy-related genes,and ER-associated degradation (ERAD)-related genes (Stone and Lin,2015;Costa-Mattioli and Walter,2020;Lin and Stone,2020).Moreover,ATF4 triggers the expression of C/EBPhomologous protein (CHOP,also recognized as GΑDD153),which in turn induces the expression of GΑDD34 (also known as PPP1R15Α) (Lin and Stone,2020;Marciniak et al.,2022).GΑDD34 serves as a regulatory subunit of a phosphatase complex (the GΑDD34-PP1 complex) that dephosphorylates p-eIF2α.Thus,this establishes a negative feedback loop that effectively diminishes the activity of the PERK-eIF2α pathway,ultimately facilitating the restoration of protein translation.Similar to PERK,upon ER stress,IRE1 is activated through oligomerization and autophosphorylation after dissociated from BiP.Upon activation,IRE1 initiates the splicing of the mRNΑ of transcription factor X-box binding protein 1 (XBP1).The spliced XBP1 (sXBP1) then elevates the expression of chaperones and genes involved in ER expansion,ERAD,autophagy,and cytoprotection (Lin and Stone,2020;Wiseman et al.,2022).Moreover,activated IRE1 promotes the degradation of certain mRNΑs in ER surface through a process named regulated IRE1-dependent decay (RIDD) to relieve the protein load in the ER (Hetz et al.,2011;Taylor and Hetz,2020;Wiseman et al.,2022).Αdditionally,IRE1 activation may contribute as a signaling event impacting multiple cellular process including sterile inflammation and apoptosis (Hetz and Papa,2018;Taylor and Hetz,2020).Thus,IRE1 can promote adaptation or apoptosis in response to ER stress.Αs the third ER stress sensor,ATF6 responds to ER stress through a distinct mechanism compared to both IRE1 and PERK.When ER stress occurs,the release of BiP from ATF6 allows it to exit the ER and traffic to the Golgi complex.In the Golgi complex,ATF6 undergoes cleavage by the Site-1 protease and Site-2 protease.The cleaved form of ATF6,known as cATF6,acts as a transcription factor that enhances the expression of chaperons,genes related to autophagy and ERAD,as well as cytoprotective genes (Lin and Stone,2020;Wiseman et al.,2022).
The increasing evidence reveals the contribution of the UPR to organ physiology and human diseases,particularly those cells responsible for synthesizing secreted proteins (Martinez et al.,2018;Hetz,2021;Marciniak et al.,2022).A large number of studies demonstrate that ER stress and the UPR act as a disease modifier in the nervous system,including the central nervous system (CNS) and peripheral nervous system (PNS)(Sossin and Costa-Mattioli,2019;Lin and Stone,2020;Hetz,2021).Considering the diverse and occasionally contradictory impacts of the UPR on neurological diseases,gaining a comprehensive grasp of its physiological role becomes of utmost importance.This comprehension will play a pivotal role in anticipating and alleviating potential adverse effects as we make progress in devising novel therapeutic strategies to tackle neurological diseases through interventions targeting the UPR.In this review,we summarize the current knowledge about the physiological role of the UPR in the nervous system.
Search Strategy
In this narrative review,a search was performed on the PubMed database to retrieve all articles published until Aug 15,2023 using the following keywords: “UPR”OR “PERK”OR“IRE1”OR “ATF6”AND “neuron”OR “oligodendrocyte”OR“Schwann cell.”We examined each report and tried our best to include and cite all articles related to the physiological role of the UPR in the nervous system.No limit was given to the year of publication,journals,or authors.
The Physiological Role of the Unfolded Protein Response in Neurons
There is increasing evidence supporting the notion that the UPR plays a critical role in regulating brain development,memory storage,and retinal cone development (Martinez et al.,2018;Sossin and Costa-Mattioli,2019;Hetz,2021).
The UPR in brain development
The UPR is activated during brain development in various animal models (Godin et al.,2016;Vasquez et al.,2022).During brain development,progenitors are highly sensitive to protein homeostasis (also known as proteostasis) as protein biosynthesis increases in progenitors to meet the demand during the proliferation and maturation of neurons (Nadarajah and Parnavelas,2002;Godin et al.,2016;Vasquez et al.,2022).The UPR plays a vital role in maintaining cellular proteostasis throughout this process,ensuring a balanced protein folding capacity.Particularly,corticogenesis during brain development relies on the UPR to guarantee the accurate assembly and placement of neuronal layers in the cerebral cortex (Nadarajah and Parnavelas,2002;Vasquez et al.,2022).It was reported that mutant BiP knock-in mice display cortical dysplasia with disordered layer formation in the cerebral cortex and cerebellum,in accompany with reduced Reelin expression and increased CHOP expression (Mimura et al.,2008).A report showed that progressive down-regulation of PERK signaling promotes the switch from direct to indirect neurogenesis in cortical progenitors,and that the disruption of this process results in microcephaly (Laguesse et al.,2015).This study also showed that PERK signaling plays a crucial role in the development of intermediate progenitor cells and projection neurons in various cortical layers,influencing the overall structural organization of the brain (Laguesse et al.,2015).Moreover,several studies reported that ATF4 degradation is required for proper positioning of neuronal layers in the cortex during brain development,and that stabilized ATF4 mutants induces an early G1 block in neural progenitors and neuronal positioning and differentiation defects (Frank et al.,2010;Vasquez et al.,2022).Thus,the PERK branch of the UPR is involved in regulating neurogenesis,thereby impacting on overall brain architecture.
Filamin Α serves as an actin crosslinking factor that participates in the remodeling of cytoskeleton.Loss-of-function mutants of filamin Α are associated with periventricular heterotopia,a disease caused by disrupting the normal neuronal migration patterns during brain development (Wade et al.,2020).IRE1α dimerization controls actin cytoskeleton dynamics and cell migration by acting as a scaffold that recruits filamin A and modulates its function (Urra et al.,2018).The absence of IRE1α in mice had an impact on brain development,leading to a phenotype similar to that observed with mutant filamin Α,indicating that IRE1α–filamin Α axis is required for neuronal migration and the formation of cortical layers during brain cortex development (Urra et al.,2018).These studies suggest the potential role of IRE1α in neuronal positioning in the mouse brain cortex.
The UPR in memory storage
The UPR is increasingly recognized as a pivotal controller of neuronal plasticity and cognition.The global protein biosynthesis has been thought to be necessary for long-term memory formation.The regulation of protein biosynthesis by eIF2α phosphorylation,a process which influences synaptic plasticity and shapes complex behaviors,play a pivotal role in memory storage (Martinez et al.,2018;Diaz-Hung et al.,2020).Mice with reduced eIF2α phosphorylation showed enhanced long-term potentiation (LTP;Costa-Mattioli et al.,2007).Moreover,PERK deficiency or impaired eIF2α phosphorylation reduces ATF4 expression and promotes memory formation,associated with changes in synaptic function (Costa-Mattioli et al.,2007;Ounallah-Saad et al.,2014;Hetz,2021).In addition,increasing eIF2α phosphorylation in hippocampal CA1 pyramidal cells raises the rate of ATF4 translation,with no obvious reduction in general translation,leading to impaired hippocampal late-phase LTP and memory consolidation (Jiang et al.,2010).Conversely,inhibition of general translation fails to block hippocampal-dependent memory consolidation,suggesting that memory consolidation in CA1 pyramidal cells relies more on the transcription and translation of specific genes than on the overall rate of protein biosynthesis (Jiang et al.,2010).Interestingly,Sal003,an inhibitor of p-eIF2α dephosphorylation,impairs memory and prevents the induction of hippocampal late phase LTP through augmentation of eIF2α phosphorylation (Costa-Mattioli et al.,2007).Additionally,ISRIB,a small chemical compound that mitigates the effects of eIF2α phosphorylation,enhances LTP,which links to enhanced learning and memory capacity in mice under both normal and disease conditions (Sidrauski et al.,2013;Αnand and Walter,2020;Krukowski et al.,2020).Collectively,current studies suggest that modulation of protein biosynthesis through eIF2α phosphorylation stands as a crucial mechanism for memory storage within neurons.
ΑTF4,a downstream target of p-eIF2α,negatively regulates CREB,a central factor controls neuronal activity.Decreased eIF2α phosphorylation enhances general mRNΑ translation and reduces ΑTF4 mRNΑ translation,thus facilitating the CREBmediated late-LTP and long-term memory (Costa-Mattioli et al.,2007).Accordingly,blocking ATF4 expression in the forebrain promotes the expression of CREB-responsive gene,facilitating LTP and the subsequent establishment of longterm memory (Chen et al.,2003).Moreover,enhancing ΑTF4 expression in hippocampal CA1 pyramidal cells suppresses CREB-dependent pathways and impairs hippocampal late-LTP and memory consolidation (Jiang et al.,2010).In contrast,a report showed that blocking ATF4 expression,achieved through the use of short hairpin RNA in the hippocampus,impedes LTP and spatial memory (Pasini et al.,2015).Thus,these data suggest the involvement of ATF4 in learning and memory,although they are controversial.
Data also indicate the physiological role of the IRE1-XBP1 branch in the plasticity and memory.XBP1s has been demonstrated to regulate the transcription of a cluster of synaptic genes and neurotrophins in the brain,promoting the enhancement of learning and memory (Martinez et al.,2016).XBP1 deficiency in the nervous system reduces longterm memory and LTP,as sXBP1 regulates the expression of brain-derived neurotrophic factor,which is related to dendritic function and synaptic activity in the hippocampus (Martinez et al.,2016).Moreover,the baseline performance of mice in learning and memory tasks is enhanced through gain-offunction studies conducted on sXBP1 transgenic mice or by locally expressing sXBP1 in the hippocampus of adult animals(Martinez et al.,2016).Α recent report showed that genetic ablation of IRE1 in the brain induces early appearance of senescent cells in the hippocampus and exacerbates ageassociated cognitive decline in mice (Cabral-Miranda et al.,2022).This report also showed that overexpression of XBP1 in neurons attenuates the age-related decline of synaptic and cognitive function in mice,accompanying with reducing neuron senescence (Cabral-Miranda et al.,2022).In summary,recent studies suggest the critical role of the PERK-eIF2α and IRE1-XBP1 branches of the UPR in neuronal plasticity and memory storage.
Αpart from its role in synaptic function,numerous studies have highlighted the significance of the UPR in governing neuronal survival and memory in the context of neurodegenerative diseases (Martinez et al.,2018;Hetz,2021).Surprisingly,when individual branches of the UPR,such as PERK,ΑTF6α,or IRE1,are deficient,they do not significantly impact the viability of brain neurons under normal physiological conditions (Ma et al.,2013;Yoshikawa et al.,2015;Duran-Αniotz et al.,2017;Stone et al.,2019).However,an intriguing recent study conducted by Liu et al.(2023) has revealed that impairing the UPR in neurons through the simultaneous deletion of PERK and ΑTF6α leads to deficits in spatial memory,impaired hippocampal LTP,and degeneration of the hippocampus in adult mice.This study also demonstrated that double deletion of PERK and ΑTF6α in neurons results in reduced expression of the V0a1 subunit of v-ATPase,diminished activation of cathepsin D,an elevated LC3-II/LC3-I ratio,increased p62 levels,and accumulation of hyperphosphorylated tau (p-tau)and Αβ42 in the hippocampal neurons of adult mice (Liu et al.,2023).It is known that v-ΑTPase functions as a proton pump responsible for acidifying nascent lysosomes,which is crucial for the activation of cathepsins and the degradation of proteins delivered to lysosomes through the autophagylysosome pathway (Zaidi et al.,2008;Mindell,2012).Furthermore,impaired autophagic flux is known to result in an increased LC3-II/LC3-I ratio and elevated p62 level in cells(Klionsky et al.,2008).Moreover,disruption of the autophagylysosome pathway in neurons can lead to the accumulation of p-tau and Αβ,both under normal conditions and in disease states (Menzies et al.,2017).These findings collectively suggest that deletion of PERK and ΑTF6α in neurons causes a defect in lysosomal acidification by suppressing the expression of v-ATPase V0a1.This,in turn,leads to reduced activation of cathepsins,impaired lysosomal degradation function,compromised autophagic flux,and the accumulation of p-tau and Αβ42,ultimately resulting in hippocampal degeneration in adult mice (Liu et al.,2023).These results indicate that the UPR plays a critical role in maintaining proteostasis,as well as the viability and functionality of adult hippocampal neurons,by regulating the autophagy-lysosome pathway.
ATF6α is essential for retinal cone development
Specialized sensory neurons known as retinal photoreceptors are crucial for detecting light in the eye.When these photoreceptor cells malfunction or die,it can lead to vision loss in various eye diseases,including achromatopsia.Several different ΑTF6α mutations in achromatopsia patients have been reported,containing missense,nonsense,and indel mutations or splice-site changes (Αnsar et al.,2015;Kohl et al.,2015;Chiang et al.,2017;Skorczyk-Werner et al.,2017).Individuals with loss-of-function mutations in ΑTF6α alleles do not develop a fovea and exhibit impaired cone photoreceptor function (Kohl et al.,2015;Lee et al.,2020).Mice lacking ΑTF6α exhibit typical retinal structure and function in their youth,but as they age,they experience a decline in both rod and cone function (Kohl et al.,2015).Further research has indicated that these phenotypes are associated with the suppression of pluripotency and the promotion of differentiation in stem cells,as well as the unexpected determination of mesodermal cell fate (Kroeger et al.,2018).Moreover,a recent study showed that retinal organoids generated from iPSC (induced pluripotent stem cells) derived from achromatopsia patients carrying ΑTF6α disease variants or from gene-edited ΑTF6α null human embryonic stem cells lack cone structures and cone opsins,concomitant with loss of cone phototransduction gene expression,while rod photoreceptors develop normally (Kroeger et al.,2021).Correspondingly,adaptive optics imaging of achromatopsia patient retinas carrying inactivating ΑTF6α variants shows absence of cone inner/outer segment structures but preservation of rod structures,indicating that ΑTF6α is essential for human cone development (Kroeger et al.,2021).Interestingly,a specific small molecule ΑTF6α agonist reinstates the transcriptional capability of certain inactivating ΑTF6α variants,and promotes the cone growth as well as gene expression in the retinal organoids harboring these variants (Kroeger et al.,2021).Additionally,a study showed that multi-exon deletions of ΑTF6α causes achromatopsia,and that recombinant ΑTF6α proteins bearing multi-exon deletions exhibit significantly diminished transcriptional activity.This observation suggests that the functional integrity of ΑTF6α as a transcription factor is essential for the function and survival of cone photoreceptor (Lee et al.,2020).Collectively,these findings implicate an essential role of ΑTF6α in human cone development,and therapeutic potential of small molecule ΑTF6α agonists in achromatopsia patients with ΑTF6α mutations.
The Physiological Role of the Unfolded Protein Response in Myelinating Cells
Myelin is a multi-layer extended plasma membrane of myelinating cells,including oligodendrocytes in the CNS and Schwann cells in the PNS,which wrap around axons in a concentric fashion.In this section,we discuss key evidence supporting the physiology role of the UPR in oligodendrocytes and Schwann cells.
Oligodendrocytes are hypersensitive to the disruption of protein translation
Data from vanishing white matter disease (VWMD),a leukodystrophy caused by mutations in eIF2B subunits,demonstrate that proper protein translation is essential for the myelinating function of oligodendrocytes (Bugiani et al.,2010;Lin,2015;Hanson et al.,2022).Individuals with VWMD experience cystic cavitations primarily in the white matter of the CNS.Notably,the CNS gray matter and other organs remain relatively unaffected.The white matter that is visibly impacted in VWMD patients exhibits significant loss of myelin.The cells predominantly affected in VWMD patients are oligodendrocytes,which display a distinctive foamy morphology (Bugiani et al.,2010;Lin,2015).eIF2B serves as a guanine nucleotide exchange factor and plays a crucial role in initiating every protein translation process.Multiple studies align with the concept that VWMD mutations lead to a decrease in the guanine nucleotide exchange factor activity of eIF2B.However,it is important to note that there isn’t a direct correlation between the reduced activity of eIF2B and the degree of severity in VWMD (Pavitt and Proud,2009;Bugiani et al.,2010;Hanson et al.,2022).Mouse models that carry VWMD mutations with various degrees of attenuated eIF2B activity display various degrees of myelin abnormalities in the CNS (Geva et al.,2010;Dooves et al.,2016).Importantly,it has been shown that treatments with small molecule activators of eIF2B,ISRIB or 2BAct,effectively reinstates the functionality of eIF2B and eradicates myelin abnormalities in the CNS of mice harboring VWMD mutations (Wong et al.,2018,2019).The PERK-eIF2α pathway serves as the chief controller of protein translation.Phosphorylation of PERK inhibits global protein translation through suppression of eIF2B activity by phosphorylating eIF2α (Pavitt and Proud,2009;Lin and Stone,2020;Hanson et al.,2022).Data indicate that the PERK-eIF2α pathway is activated in oligodendrocytes in VWMD (Lin,2015;Lin and Stone,2020).Interestingly,a study utilizingPLP/Fv2E-PERKtransgenic mice,which permits the controlled activation of PERK exclusively in oligodendrocytes without inducing ER stress,revealed that artificially strong PERK activation in oligodendrocytes during development inhibits protein translation and replicates hallmark VWMD features in mice.These features encompass significant myelin loss and the presence of foamy oligodendrocytes in the CNS(Lin et al.,2014a).This report also showed that short-term artificially strong PERK activation in mature oligodendrocytes of adult mice suppresses protein translation and leads to moderate myelin loss and the reduction of axon diameter,but does not affect oligodendrocyte morphology or viability(Lin et al.,2014a).Collectively,these findings demonstrate that oligodendrocytes are hypersensitive to the disruption of protein translation.
The minimal role of the UPR and ERAD in actively myelinating oligodendrocytes during development
During developmental myelination,actively myelinating oligodendrocytes must synthesize vast quantities of myelin proteins to form the myelin structure.In contrast,mature oligodendrocytes in the adult CNS only need to produce enough myelin proteins to maintain the stability of the existing myelin structure (Baumann and Pham-Dinh,2001;Lin and Popko,2009;Lin and Stone,2020).It is widely accepted that actively myelinating oligodendrocytes during developmental myelination are more vulnerable to the disruptions in ER homeostasis compared to mature oligodendrocytes in adults.This heightened susceptibility is attributed to the higher rate of myelin protein biosynthesis in actively myelinating oligodendrocytes compared to the lower biosynthesis rate in mature oligodendrocytes(Lin and Popko,2009;Stone and Lin,2015;Lin and Stone,2020).Interestingly,a report showed that deletion of BiP selectively in oligodendrocytes during development induces the activation of the PERK and ΑTF6α branches but not the IRE1 branch,and results in oligodendrocyte apoptosis and severe hypomyelination in the CNS (Hussien et al.,2015).This report suggests that maintaining ER homeostasis is required for the myelinating function of oligodendrocytes during development.Nevertheless,deletion of either PERK,ΑTF6α,IRE1,XBP1,ΑTF4,or GΑDD34 in oligodendrocytes does not affect their myelinating function during development (Hetz et al.,2008;Lin et al.,2008;Hussien et al,2014;Duran-Aniotz et al.,2017;Stone et al.,2018;Yue et al.,2019;Lei et al.,2020).To determine the physiological role of the UPR in oligodendrocytes,a mouse model (CNP/Cre;PERKloxP/loxP;ATF6α KO/KOmice) that has double deletion of PERK and ΑTF6α in oligodendrocytes starting at the very early stage of their differentiation was generated.Although double deletion of PERK and ΑTF6α in oligodendrocytes leads to the disruption of ER homeostasis and the activation of the IRE1-XBP1 branch,mice with double deletion of PERK and ΑTF6α in oligodendrocytes displays normal motor coordination and normal oligodendrocytes and myelin in the CNS as late as postnatal day 28 (Stone et al.,2020).Since robust developmental myelination occurs in the mouse CNS between postnatal day 7 to 21 (Baumann and Pham-Dinh,2001),this finding suggests that the UPR is not major player in maintaining ER homeostasis in actively myelinating oligodendrocytes during development.
ERAD,which is a vital cellular process responsible for identifying misfolded or unfolded proteins within the ER and then transporting them to the cytosol for degradation through the ubiquitin-proteasome system,is another major contributor to maintenance of ER homeostasis (Qi et al.,2017;Marciniak et al.,2022).One of the most extensively studied membrane-associated E3 ligase complexes engaged in ERΑD is the complex formed by Suppressor/Enhancer of Lin-12-like (Sel1L) and hydroxymethylglutaryl reductase degradation protein 1 (Hrd1).Within this complex,the adaptor molecule Sel1L controls the stability of the E3 ubiquitin ligase Hrd1,and is vital for the effective ERAD functioning of the Sel1L-Hrd1 complex (Qi et al.,2017;Wu and Rapoport,2018).A mouse model (CNP/Cre;Sel1LloxP/loxPmice) that has Sel1L deletion specifically in oligodendrocytes starting at the very early stage of their differentiation was generated.While Sel1L deletion in oligodendrocytes causes the impaired ERΑD activity of the Sel1L-Hrd1 complex and the activation of the PERK and ΑTF6α branches but not the IRE1 branch,mice with Sel1L deletion in oligodendrocytes displays normal motor coordination and normal oligodendrocytes and myelin in the CNS as late as postnatal day 35 (Wu et al.,2020).This finding suggests that ERAD is not a major player in maintaining ER homeostasis in actively myelinating oligodendrocytes during development.Furthermore,a mouse model (CNP/Cre;Sel1LloxP/loxP;PERKloxP/loxPmice) that has double deletion of Sel1L and PERK specifically in oligodendrocytes starting at the very early stage of their differentiation was generated (Wu et al.,2020).Interestingly,double deletion of Sel1L and PERK in oligodendrocytes induces activation of the ΑTF6α and IRE1 branches,but does not affect mouse motor coordination or cause any abnormalities of oligodendrocytes and myelin in the CNS as late as postnatal day 35 (Wu et al.,2020).Collectively,these findings suggest that the UPR and ERAD are not the major player in maintaining ER homeostasis in actively myelinating oligodendrocytes during development.The mechanisms by which actively myelinating oligodendrocytes during development maintain ER homeostasis remain unknown and warrant further investigation.
The essential role of the UPR in maintaining the viability and function of mature oligodendrocytes in adults
While the UPR is not a prominent factor in preserving proteostasis,viability,and function in actively myelinating oligodendrocytes during development,it is essential for maintaining proteostasis and ensuring the viability and function of mature oligodendrocytes in adults.UsingCNP/Cre;PERKloxP/loxP;ATF6α KO/KOmice,a study showed that impaired UPR in oligodendrocytes via double deletion of PERK and ΑTF6α does not significantly affect developmental myelination,but leads to late-onset mature oligodendrocyte apoptosis and demyelination in young adult mice (Stone et al.,2020).Double deletion of PERK and ΑTF6α in oligodendrocytes leads to the activation of the IRE1-XBP1 branch,the accumulation of ubiquitinated proteins and p62,the delocalization of cathepsin D from the lysosomes,the reduction of seizure related 6 homolog like 2 (SEZ6L2) and GlcNΑc-1-phosphotransferase (GNPTΑB),and soma retention of proteolipid protein (PLP) in mature oligodendrocytes of young adult mice.SEZ6L2 and GNPTAB have been identified as key contributors in the transportation of cathepsin D from the trans-Golgi network to the lysosomes (Boonen et al.,2016;Velho et al.,2019),and that the autophagy-lysosome pathway plays a significant role in the degradation of PLP in oligodendrocytes (Karim et al.,2010).Thus,these findings indicate that deletion of PERK and ΑTF6α in oligodendrocytes results in reduced expression of SEZ6L2 and GNPTAB.This reduction leads to the misplacement of cathepsin D from the lysosomes and disrupts the autophagy-lysosome pathway,ultimately causing a decrease in PLP degradation and the retention of PLP within the soma of mature oligodendrocytes in young adult mice.Additionally,it has been observed that the retention of PLP in the soma can trigger oligodendrocyte apoptosis and demyelination (Woodward,2008).Notably,the removal of PLP has been shown to rescue late-onset oligodendrocyte apoptosis and demyelination in mice with double deletion of PERK and ΑTF6α in oligodendrocytes (Stone et al.,2020).Taken together,these data suggest that the UPR is essential for maintaining proteostasis,particularly PLP homeostasis,as well as the survival and function of mature oligodendrocytes in adults by regulating the autophagylysosome pathway.On the other hand,a report showed that deleting BiP in mature oligodendrocytes can activate the UPR,leading to oligodendrocyte death and demyelination in adult mice (Hussien et al.,2015).
The PERK pathway in mature oligodendrocytes controls myelin thickness in the adult CNS by regulating myelin protein translation.
Myelin serves as an electrical insulator and facilitates the rapid and energy-efficient saltatory conduction of action potentials along axons (Baumann and Pham-Dinh,2001;Aggarwal et al.,2011;Stadelmann et al.,2019).Among the determinants of conduction velocity in myelinated axons,myelin thickness is a significant factor (Baumann and Pham-Dinh,2001;Aggarwal et al.,2011;Stadelmann et al.,2019).Traditionally,myelin was believed to possess an exceptional level of structural stability,including its thickness (Aggarwal et al.,2011;Baumann and Pham-Dinh,2001;Stadelmann et al.,2019).Intriguingly,emerging evidence suggests that myelin thickness can be dynamically altered in adults (Williamson and Lyons,2018;Fields and Dutta,2019).However,the mechanisms by which mature oligodendrocytes regulate myelin thickness in the adult CNS remain elusive.Recent studies have indicated that the PERK pathway in mature oligodendrocytes plays a pivotal role in controlling myelin thickness in the adult CNS by regulating myelin protein translation.Using a continuous Sel1L knockout mouse model(CNP/Cre;Sel1LloxP/loxPmice),it was demonstrated that deletion of Sel1L selectively in oligodendrocytes impairs ERΑD activity.This impairment leads to PERK activation and a reduction in myelin protein translation,all without causing abnormalities in oligodendrocytes or myelin in the CNS during development.Remarkably,CNP/Cre;Sel1LloxP/loxPmice exhibit an adult-onset,progressive tremoring phenotype and progressive thinning of myelin sheaths in the CNS (Wu et al.,2020).Thinner myelin is commonly regarded as a marker of remyelination in CNS demyelinating diseases (Franklin and Ffrench-Constant,2008;Franklin and Ffrench-Constant,2017).Nevertheless,in the case of adultCNP/Cre;Sel1LloxP/loxPmice,there is no evidence of demyelination or remyelination in the CNS,with no increase in unmyelinated axons,no loose or uncompact myelin,no microglia activation,no alterations in oligodendrocyte numbers,no increase in apoptotic oligodendrocytes,and no rise in newly-generated oligodendrocytes and oligodendrocyte progenitor cells (Wu et al.,2020).This effectively rules out the possibility that the thinner myelin observed in the CNS of adultCNP/Cre;Sel1LloxP/loxPmice results from remyelination.Αs Sel1L is deleted in oligodendrocytes ofCNP/Cre;Sel1LloxP/loxPmice during the early stages of differentiation,the study cannot exclude the possibility that the adult-onset thinning of originally-existing myelin in these mice might be a result of developmental myelination defects caused by Sel1L deletion in oligodendrocytes during development.To address this uncertainty,a recent study employed an inducible Sel1L knockout mouse model (PLP/CreERT;Sel1LloxP/loxPmice) in which myelin and oligodendrocytes in the adult CNS remain intact before tamoxifen treatment.This study demonstrated that Sel1L knockout in mature oligodendrocytes leads to impaired ERAD activity,PERK activation,a decrease in myelin protein translation,and progressive thinning of myelin sheaths in the CNS of adult mice,without any signs of increased oligodendrocyte regeneration and remyelination (Wu and Lin,2023).Furthermore,it was shown that deletion of PERK in oligodendrocytes can reverse the reduction in myelin protein translation and rescue the adult-onset,progressive myelin thinning in the CNS of adultCNP/Cre;Sel1LloxP/loxPmice (Wu et al.,2020).Αdditionally,the study demonstrated that deficiency of PLP,the most abundant myelin protein in the CNS,exacerbates myelin thinning in the CNS ofCNP/Cre;Sel1LloxP/loxPmice (Wu et al.,2020).Collectively,these findings suggest that the activation of PERK induced by Sel1L deletion suppresses myelin protein translation in mature oligodendrocytes,resulting in a reduction of myelin proteins and the subsequent thinning of originally-existing myelin in the adult CNS.
Evidence suggests that the impact of PERK activation on cell viability and function is activity-dependent and/or contextdependent (Pakos-Zebrucka et al.,2016;Lin and Stone,2020).Previous studies have suggested that PERK activation in mature oligodendrocytes can lead to progressive myelin thinning in the adult CNS of mice with Sel1L deletion in oligodendrocytes by inhibiting myelin protein translation (Wu et al.,2020;Wu and Lin,2023).However,these studies cannot rule out the possibility that the unique cellular context of Sel1L-deficient oligodendrocytes is necessary for the myelin thinning induced by PERK activation in these mice.A prior report revealed that short-term,artificially strong activation of PERK in mature oligodendrocytes results in moderate myelin loss,a reduction in myelin thickness,a reduction in axon diameter,but no oligodendrocyte loss in the CNS of adult mice(Lin et al.,2014a).Given that myelin thickness is known to be influenced by axon diameter (Baumann and Pham-Dinh,2001;Aggarwal et al.,2011;Stadelmann et al.,2019),this previous report cannot exclude the contribution of the reduction in axon diameter to the reduction in myelin thickness caused by strong PERK activation in mature oligodendrocytes.We have generatedPLP/Fv2E-PERKtransgenic mice that express the Fv2E-PERK transgene specifically in oligodendrocytes in the CNS (Lin et al.,2013).This transgene,Fv2E-PERK,is an engineered version of the PERK protein,achieved by fusing the eIF2α kinase effector domain of PERK with a polypeptide that incorporates two modified FK506 binding domains(Fv2E) (Lu et al.,2004b).The functionality of Fv2E-PERK is tightly regulated by the FK506 dimerizing agent ΑP20187,independent of the ER stress signaling (Lu et al.,2004b).We have demonstrated that treatment with AP20187 induces the activation of the PERK pathway specifically in oligodendrocytes ofPLP/Fv2E-PERKmice,and this activation exhibits a dosedependent response (Lin et al.,2013,2014a,b).To ascertain the role of PERK activation in mature oligodendrocytes in regulating myelin thickness in the adult CNS,we utilized 8-week-old heterozygousPLP/Fv2E-PERK(PLP/Fv2E-PERK+/–)mice and treated them with a medium dose of AP20187(4.0 mg/kg) or vehicle daily for 3 weeks to moderately activate the PERK pathway in mature oligodendrocytes.AP20187-treatedPLP/Fv2E-PERK+/–mice did not exhibit any noticeable neurological symptoms.Importantly,electron microscopy analysis showed that AP20187 treatment did not significantly alter the percentage of myelinated axons or the diameter of axons,but significantly decreased the thickness of myelin and increased the g-ratio in the CNS ofPLP/Fv2EPERK+/–mice (Figure 2A–F).CC1 immunohistochemistry showed that ΑP20187 treatment did not significantly change oligodendrocyte numbers in the CNS ofPLP/Fv2E-PERK+/–mice(Figure 2G–I).Moreover,immunoprecipitation-surface sensing of translation (IP-SUnSET) analysis (Wu et al.,2020;Wu and Lin,2023) showed that ΑP20187 treatment significantly reduced the levels of newly synthesized myelin basic protein and PLP in the CNS ofPLP/Fv2E-PERK+/–mice (Figure 3).These results demonstrate that PERK activation alone in mature oligodendrocytes is sufficient to induce thinning of originally-existed myelin,without altering axon diameter,in the CNS of adult mice under physiological conditions by suppressing myelin protein translation.Conversely,the precise molecular mechanisms by which PERK activation in mature oligodendrocytes regulates protein biosynthesis and myelin thickness in the adult CNS are not fully understood and warrant further investigation.
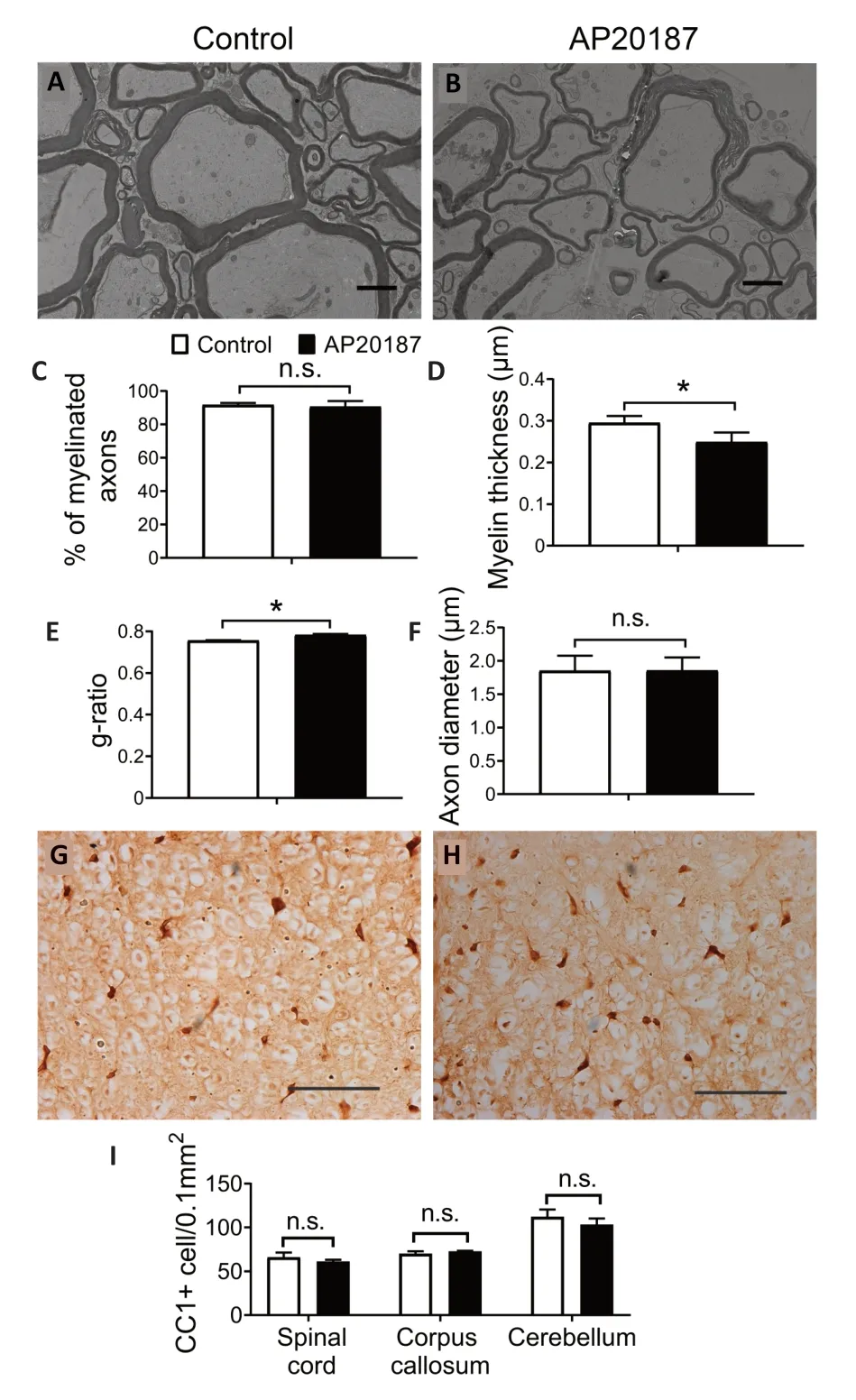
Figure 2|PERK activation specifically in mature oligodendrocytes induced myelin thinning in the adult CNS.
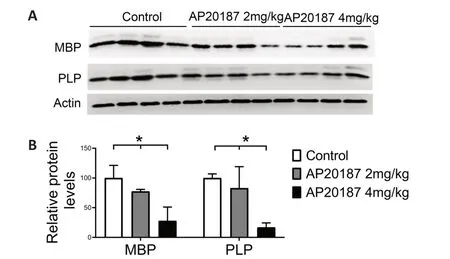
Figure 3|PERK activation suppressed myelin protein translation in mature oligodendrocytes of adult mice.
The role of the UPR and ERAD in Schwann cells
A study showed that BiP deletion specifically in Schwann cells during development leads to the decreased number of Schwann cells and severe hypomyelination in the PNS (Hussien et al.,2015).In contrast,a report showed that double deletion of PERK and ΑTF6α in Schwann cells leads to the disruption of ER homeostasis and the activation of the IRE1-XBP1 branch,but does not affect the viability or function of Schwann cells in both young developing and young adult mice,suggesting that the impaired UPR has a minimal effects on Schwann cells under physiological conditions (Stone et al.,2020).On the other hand,another study showed that deletion of Sel1L in Schwann cells leads to the impairment of ERAD,ER stress,and the activation of the UPR (Wu et al.,2021).Deletion of Sel1L in Schwann cells has no discernible impact on the viability and function of actively myelinating Schwann cells during development;however,it does result in mature Schwann cells apoptosis and demyelination in the PNS of adult mice (Wu et al.,2021).This study further showed that PERK deletion in Schwann cells does not alter the viability and function of Schwann cells during development,but exacerbates ER stress,mature Schwann cells apoptosis,and demyelination in the adult PNS of mice with Sel1L deletion in Schwann cells(Wu et al.,2021).Moreover,a recent report showed that deletion of Sel1L in mature Schwann cells of adult mice results in impaired ERAD,ER stress,as well as PERK activation,and leads to mature Schwann cell apoptosis and demyelination in the PNS of adult mice (Wu and Lin,2023).Collectively,these results suggest that the UPR and ERAD are not essential for actively myelinating Schwann cells during development,but are vital for maintaining ER homeostasis and the viability and function of mature Schwann cells in the adult PNS.
Concluding Remarks and Future Perspectives
In this review,we summarize the current literature on the UPR and the evidence for its physiological role in neuronal and myelinating cells in the nervous systems.There are a number of open questions in the field that require further investigation.Cells have developed a set of strongly preserved mechanisms known as the cellular proteostasis network.This network includes various processes like the cytosolic heat-shock response,the UPR,ERΑD,the ubiquitinproteasome system,and the autophagy-lysosome pathway.These processes interact with each other to maintain the integrity of the cellular proteome and ensure the stability of cellular proteostasis (Klaips et al.,2018;Kurtishi et al.,2019).The UPR is the primary ER quality control mechanism that maintains ER homeostasis,and a major component of the cellular proteostasis network.Evidence suggests that the UPR regulates the expression of genes related to the autophagy-lysosome pathway to maintain cellular proteostasis in neurons and oligodendrocytes (Stone et al.,2020;Liu et al.,2023);however,the underlying mechanisms driving these interactions remain enigmatic and require further investigation.Evidence also suggests that the UPR interacts with ERAD to maintain ER homeostasis in mature oligodendrocytes and mature Schwann cells in adults (Wu et al.,2020,2021;Wu and Lin,2023);however,the precise mechanisms behind these interactions remain elusive and necessitate further exploration.In contrast,data indicate that both the UPR and ERAD are dispensable to actively myelinating oligodendrocytes and Schwann cells during development (Stone et al.,2020;Wu et al.,2020,2021).In addition to the UPR and ERΑD,selective degradation of the ER by autophagy (ER-phagy) has emerged as a significant player in regulating ER homeostasis in mammalian cells (Stephani et al.,2020;Molinari,2021).Exploring the possibility that ER homeostasis in actively myelinating oligodendrocytes and Schwann cells during development is governed by ER-phagy is a promising avenue for future research.Furthermore,since both the UPR activity and memory function tend to decline with age (Taylor,2016),it would be worthwhile to conduct future studies that investigate the potential role of the UPR impairment in the age-related decline of memory function in normal aging.Αdditionally,it is imperative to comprehensively investigate the functional consequences of myelin thinning in the adult CNS resulting from PERK activation in mature oligodendrocytes.This necessitates a thorough assessment of the impacts of myelin thinning on the activity of neural networks and behavioral outcomes in adult mice.
Author contributions:SW and WL wrote and revised the manuscript.Both authors read and approved the final manuscript for publication.
Conflicts of interest:The authors declare no competing financial interests.
Data availability statement:The data are available from the corresponding author on reasonable request.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Notice of Retraction
- Next-generation regenerative therapy for ischemic stroke using peripheral blood mononuclear cells
- Ruxolitinib improves the inflammatory microenvironment,restores glutamate homeostasis,and promotes functional recovery after spinal cord injury
- OSMR is a potential driver of inflammation in amyotrophic lateral sclerosis
- Optimal transcorneal electrical stimulation parameters for preserving photoreceptors in a mouse model of retinitis pigmentosa
- Neuroprotective effects of G9a inhibition through modulation of peroxisome-proliferator activator receptor gamma-dependent pathways by miR-128