基于生物信息学的子宫内膜癌预后模型构建
2024-03-04林鹏孙培许淑霞
林鹏 孙培 许淑霞


[摘要] 目的 筛选与子宫内膜癌(endometrial carcinoma,EC)预后相关的差异基因并构建预后模型。方法 从癌症基因图谱(The Cancer Genome Atlas,TCGA)数据库和基因表达谱数据库(Gene Expression Omnibus,GEO)的数据集GSE63678中下载EC和正常对照样本的基因表达数据,筛选出共有差异基因。采用LASSO回归分析筛选出具有预后意义的基因,并构建预后特征。从TCGA数据库中获取具有完整信息的EC患者,按1∶1的比例随机分为训练组和验证组。對训练组患者基于预后特征构建生存曲线;采用基因本体论(gene ontology,GO)分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)对预后特征进行功能注释和通路富集分析;结合预后特征及临床危险因素构建列线图,采用校准曲线和C指数评估列线图性能。最后使用验证组加以验证。结果 从TCGA和GEO数据库分别筛选出4800个和257个差异基因,其中共表达的上调基因73个,下调基因52个;LASSO回归分析筛选出6个预后基因,分别为ORMDL2、BNC2、TTK、MAMLD1、KCTD7、DCLK2;生存曲线结果表明高风险组患者的总生存率显著低于低风险组(P<0.01);GO分析和KEGG结果显示预后特征与细胞周期相关。列线图在训练组与验证组中均显示出良好的预测能力。结论 本研究构建一种基于预后特征的预测模型,可准确预测EC患者的预后,为临床诊疗提供新的理论支持。
[关键词] 子宫内膜癌;生物信息学;预后;预测模型
[中图分类号] R737.3 [文献标识码] A [DOI] 10.3969/j.issn.1673-9701.2024.03.011
Construction of prognostic model for endometrial carcinoma based on bioinformatics
LIN Peng1, SUN Pei2, XU Shuxia2
1.Department of Pathology, Fujian Children’s Hospital (Fujian Branch of Shanghai Children’s Medical Center), College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, Fujian Fuzhou, 350000, China; 2.Department of Pathology, Fujian Maternity and Child Health Hospital, College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, Fuzhou 350000, Fujian, China
[Abstract] Objective Differential genes related to prognosis of endometrial carcinoma (EC) were screened and prognostic models were constructed. Methods Gene Expression data of EC and normal control samples were downloaded from The Cancer Genome Atlas (TCGA) database and Gene Expression Omnibus (GEO) dataset GSE63678 to screen out common differential genes. LASSO regression analysis was used to screen out the genes with prognostic significance and construct prognostic characteristics. EC patients with complete information were obtained from the TCGA database and randomly divided into the training group and the validation group in a ratio of 1:1. In the training group, survival curves were constructed based on prognostic characteristics. Functional annotation and pathway enrichment analysis were conducted using gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. Combined with prognostic features and clinical risk factors, a calibration curve and C-index were used to evaluate the performance of the histogram. Finally, use a verification group for validation. Results A total of 4800 and 257 differentially expressed genes were screened from TCGA and GEO databases respectively, of which 73 up-regulated genes and 52 down-regulated genes were co-expressed. 6 prognostic genes (ORMDL2, BNC2, TTK, MAMLD1, KCTD7 and DCLK2) were screened out by LASSO regression analysis. The survival curve showed that the overall survival of patients in the high-risk group was significantly lower than that in the low-risk group (P<0.01). GO analysis and KEGG results exhibited that prognostic signature was associated with cell cycle. The nomogram showed powerful predictive ability in the training and validation groups. Conclusion We constructed a predictive model based on prognostic genes, which can accurately predict the prognosis of patients with EC and provide new theoretical support for clinical diagnosis and treatment.
[Key words] Endometrial carcinoma; Bioinformatics; Prognosis; Predictive model
子宫内膜癌(endometrial carcinoma,EC)是发达国家最常见的妇科恶性肿瘤之一,发病率每年稳定增长约1.3%[1-2]。我国的发病数也在增加,尤其是在年轻女性中[3]。EC患者一般预后较好,总生存率在80%左右,其预后因素主要包括年龄、疾病的分期和组织学类型等[4]。国际妇产科联盟(Federation International of Gynecology and Obstetrics,FIGO)Ⅰ~Ⅱ期EC患者的5年生存率为74%~91%,而FIGO Ⅲ~Ⅳ期的患者5年生存率为20%~57%[5]。手术和辅助治疗可显著提高EC患者的生活质量,这些治疗也可显著延长患者的生存时间。然而,对于晚期及复发转移患者的治疗方法有限,缺乏有效的预后评估方法,导致患者预后不佳甚至死亡[6]。目前,用于评估EC患者的预后主要依靠临床分期和术后的病理分期,但因其局限于肿瘤解剖范围的分期,难以准确预测EC患者的预后[7]。因此,需要挖掘新的生物标志物并构建更加可靠的预测模型,来提高EC患者的生存率。本研究收集癌症基因图谱(The Cancer Genome Atlas,TCGA)数据库中EC患者的测序、临床和长期随访数据及基因表达谱数据库(Gene Expression Omnibus,GEO)中EC数据集GSE63678的测序数据,为EC个体化预测模型构建提供可能性。
1 材料与方法
1.1 研究对象与数据的收集
从TCGA数据库下载553个EC组织和23个正常对照样本的基因表达数据及对应的553例患者的临床数据,将具有完整可用的基因表达数据和临床信息的541例患者纳入后续分析。从GEO数据库下载数据集GSE63678的基因表达信息,包括7个EC组织样本及5个正常组织样本。
1.2 差异基因的筛选
分析从TCGA和GEO数据库中获取的基因表达数据,筛选出EC差异基因。本研究的差异基因筛选标准:若EC组织基因表达量与正常对照样本基因表达量的倍性变化(fold change,FC)>1,且P<0.001,則为上调基因;若FC<1,且P<0.001,则为下调基因。使用R语言的ggplot2包绘制差异基因火山图,并用韦恩图筛选交叉差异表达的基因。
1.3 预后模型的构建
采用R语言的glmnet和survival包对差异基因进行LASSO回归分析,将筛选出的非零系数基因视为与患者预后相关的基因。将具有完整基因表达和临床信息的541例患者按1∶1的比例随机分为训练组(n=270)和验证组(n=271),使用筛选出的预后基因及其相应的λ值计算每组患者的风险评分(风险评分=表达值ORMDL2×λORMDL2+表达值BNC2× λBNC2+表达值TTK×λTTK+表达值MAMLD1×λMAMLD1+表达值KCTD7×λKCTD7+表达值DCLK2×λDCLK2)。以训练组患者风险评分的平均数作为临界值,分为低危组和高危组。采用Kaplan-Meier方法绘制两组患者生存曲线。
1.4 功能富集分析
通过Pearson相关分析筛选出与风险评分最相关的基因(Cor>0.5,P<0.05),并将其上传至DAVID数据库中进行生物功能注释和通路富集分析。本研究选择官方的基因符号作为标识符,并选择智人作为物种,最后获得GO分析和KEGG通路分析的富集结果。本研究显示P值按升序排列的前5个结果(P<0.05)。
1.5 列线图建立
采用R语言中的Rms包进行列线图的建立与分析,上部为评分系统,下部为预测系统。EC患者的1、2、3、5、10年生存率和复发率通过总分、各因素的总和来准确预测,并使用校准曲线和C指数值显示生存预测的准确性。
1.6 统计学方法
使用R4.2.2和SPSS 26.0软件进行统计分析。总生存期为自确诊到死亡或末次随访的时间。风险评分与总生存期的关系用Kaplan-Meier生存曲线分析。在DAVID数据库上进行GO分析和KEGG通路分析。采用t检验分析风险评分与临床病理特征之间的关系。P<0.05为差异有统计学意义。
2 结果
2.1 差异基因的筛选
通过对TCGA数据进行处理分析,筛选出4800个差异基因,其中上调基因910个(19%),下调基因3890个(81%),见图1A;对GEO数据进行处理分析,筛选出257个差异基因,其中上调基因153个(60%),下调基因104个(40%),见图1B。用韦恩图取交集获得共同表达的上调差异基因73个,见图1C,共同表达的下调差异基因52个,见图1D。
2.2 LASSO回归筛选
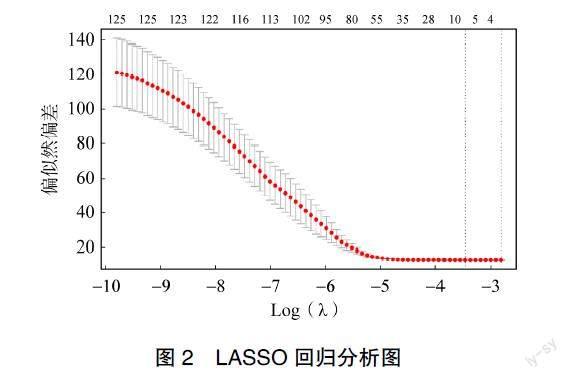
将筛选出的125个差异基因进行LASSO回归分析,见图2,最后筛选出6个非零系数的基因及其λ值(ORMDL2:‒0.0000189298687888065;BNC2:0.0000890777237690712;TTK:0.00013570127002083;MAMLD1:0.000199877136208518;KCTD7:0.000275026231175745;DCLK2:0.0003987957428 90722)。根据每个基因的表达值及其相应的λ值计算每例患者的风险评分,以训练组中风险评分的平均数(0.2845)作为临界值。
2.3 风险因素与EC患者的预后关系
在热图中,根据不同的风险水平,显示训练组和验证组中每例患者的临床特征及预后基因的表达情况,见图3A、3B。生存曲线图显示,在训练组中高风险组患者的总生存率显著低于低风险组(P<0.01),见图3C;在验证组中,得到了一致的结果(P<0.01),见图3D。
2.4 风险评分与临床因素的关系
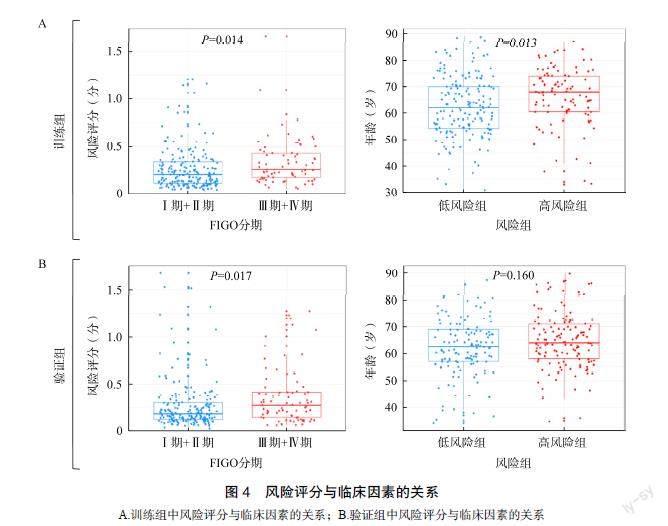
进一步检测风险评分与临床因素之间的关系。在训练组中,FIGO Ⅳ~Ⅳ期患者的风险评分显著高于FIGO Ⅰ~Ⅱ期患者(P<0.05),高风险组患者年龄显著高于低风险组患者(P<0.05),见图4A。在验证组中,FIGO Ⅳ~Ⅳ期患者的风险评分也显著增加(P<0.05),然而高风险组患者年龄与低风险组患者年龄差异无统计学意义(P>0.05),见图4B。
2.5 风险评分与细胞周期密切相关
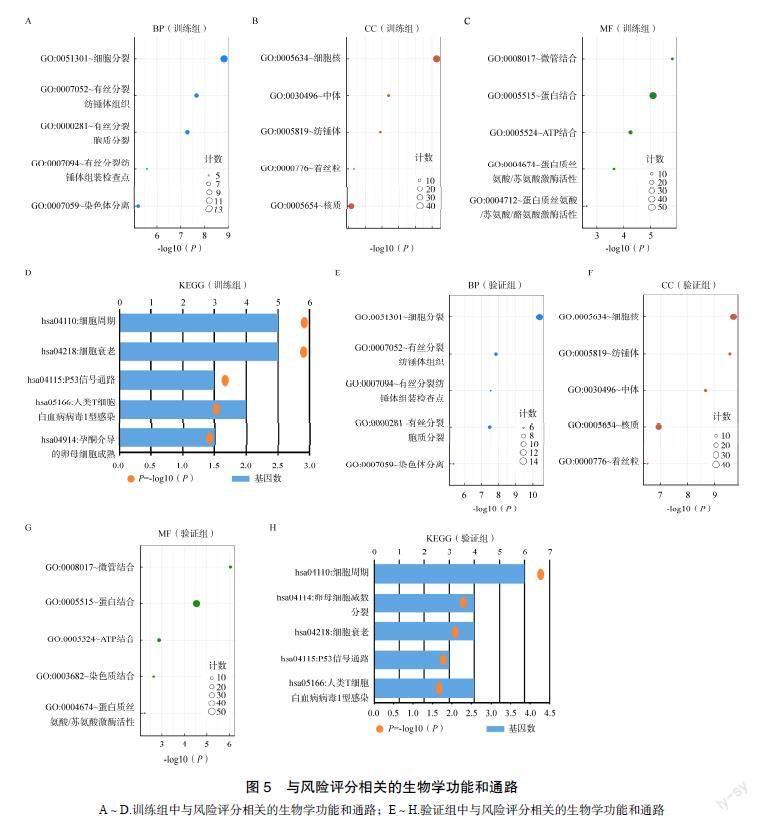
为探讨与风险评分相关的生物学功能,采用Pearson相关分析筛选出与风险评分最相关的基因(Cor>0.5,P<0.05),将上述基因进行功能富集分析。GO分析结果显示,在训练组和验证组中,与风险评分最为相关的生物过程(biological process,BP)为细胞分裂,见图4A、4E,细胞成分(cell component,CC)是细胞核,见图4B、4F,分子功能(molecular function,MF)为微管结合,见图4C、4G。KEGG结果显示,在两组数据库中,风险评分均与细胞周期最相关,见图4D、4H。
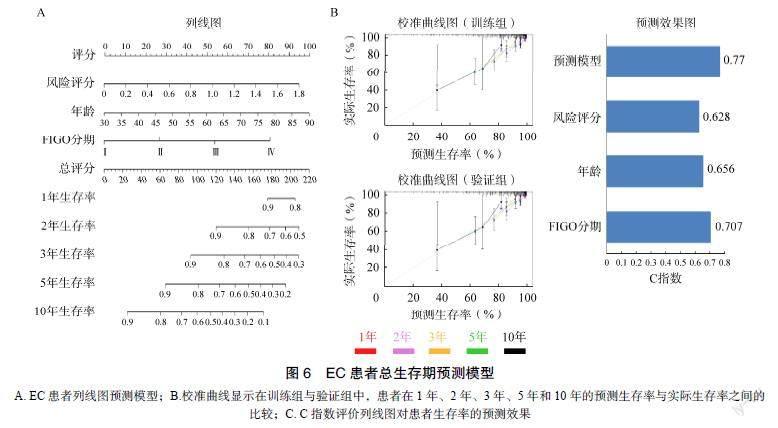
2.6 个性化预测模型具有较好的预测精度
列线图在临床实践中可用于计算特定肿瘤患者的生存率,具有重要的应用价值。因此,本研究在训练组中基于风险评分、年龄、FIGO分期等独立预测因素,构建个体化预测模型。通过该模型估计EC患者(1、2、3、5、10年)的总生存率,见图6A。校准曲线结果显示,在训练组与验证组中A和B.按风险评分的升序,热图显示训练组与验证组中每例患者的临床因素和6个风险基因的表达;C和D.训练组与验证组中高风险患者与低风险患者总生存率曲线分析图A.训练组中风险评分与临床因素的关系;B.验证组中风险评分与临床因素的关系
A~D.训练组中与风险评分相关的生物学功能和通路;E~H.验证组中与风险评分相关的生物学功能和通路
患者在1、2、3、5、10年的预测生存率接近实际生存率,说明列线图显示出良好的预测能力,见图6B。该列线图模型的C指数为0.77,优于其他任何独立的预测模型,也显示出较好的预测精度,见图6C。
3 讨论
早期EC患者由于手术等综合治疗,预后良好;然而,当EC进展为晚期或转移后,其预后很差。恶性肿瘤具有异质性,同一期患者的生存预后有所不同。虽然目前对特异性分子标志物的广泛研究取得进展,但由于个体差异,在临床实践中还没有建立准确的方法来判断患者的预后[8]。因此,迫切需要寻找更有价值的生物标志物来准确预测EC患者的预后,并提高患者的总生存期。
本研究中,笔者在TCGA和GEO数据库中筛选出共有的EC组织与正常对照样本的差异基因,并采用LASSO回归分析筛选出6个与EC患者总生存期相关的预后基因。由于GEO数据库缺乏关于EC患者的临床信息,因此本研究将整个具有完整基因表达和临床信息的TCGA患者队列随机分为训练组和验证组。然后,在训练组中构建一个基于预后特A. EC患者列线图预测模型;B.校准曲线显示在训练组与验证组中,患者在1年、2年、3年、5年和10年的预测生存率与实际生存率之间的比较;C. C指数评价列线图对患者生存率的预测效果
征的生存曲线,结果表明高风险组患者的总生存率显著低于低风险组,并在验证组中得到证实,保证了其准确性。因此,生存曲线结果说明预后特征对EC患者的预后具有独立的预测能力。
在本模型的6个预后基因中(ORMDL2、BNC2、TTK、MAMLD1、KCTD7及DCLK2),目前还无关于KCTD7在肿瘤中的功能或机制的报道,其他5个基因在各种肿瘤中的重要作用都已有报道。ORMDL2是鞘脂代谢的重要调控因子,与细胞的生长、增殖、迁移和侵袭有关。有研究表明,ORMDL2的高表达与胶质瘤的不良预后相关[9]。BNC2在各种类型的细胞中广泛存在,该基因与角质细胞增殖能力有关,在细胞终极分化中起重要作用,有研究表明BNC2不仅可抑制卵巢癌的發展,还与胶质母细胞瘤患者的生存相关[10-12]。 TTK是纺锤体组装检查点的核心组成部分,其功能是确保染色体正确分配给子细胞,许多研究表明TTK在几种人类恶性肿瘤中过度表达,包括胃癌、卵巢癌和乳腺癌,其表达与患者不良预后有关[13-15]。
MAMLA1是一个转录共激活因子,目前的研究表明MAMLA1与促性腺激素垂体腺瘤和幕上室管膜瘤的发生、发展有关[16-17]。DCLK2是一种参与神经发育和成熟的微管相关蛋白激酶,有研究表明DCLK2促进乳腺癌细胞侵袭转移,其在乳腺癌中的高表达与患者不良预后相关[18]。另有学者发现,DCLK2的高表达与慢性淋巴细胞白血病患者的生存率降低有关[19]。因此,本研究中确定的预后基因可能是靶向治疗EC的有效途径。为进一步挖掘和探索这些预后基因在EC中的功能和机制,本研究对此进行功能注释和通路富集分析。GO分析和KEGG结果显示,与风险评分最相关的基因主要富集于细胞分裂、有丝分裂、染色体分裂等生物学过程中及与细胞周期相关的信号通路中。此前有研究表明,高度有序的细胞周期活动是细胞维持正常代谢与增殖的保障,细胞周期调控异常是促进肿瘤恶性进展的潜在诱变因素[20]。早期有学者指出G2细胞周期调节因子的改变可导致子宫内膜的癌变[21]。因此,细胞周期异常在EC的发生、发展中起重要作用。
最后,为能更直观地显示EC患者的预后情况,本研究基于预后特征和其他两个独立的临床特征构建列线图,以帮助临床医生个体化预测EC患者1、2、3、5、10年的总生存率;校准曲线在训练组和验证组中均显示EC患者的预测生存率与实际生存率一致,C指数结果也表明该列线图具有良好的预测性能。因此,以上结果表明该模型可对EC患者的生存和预后进行个性化、准确的评估,有利于患者的生存和预后。本研究存在局限性,首先有限的样本量可能影响模型的精度;其次,该模型的参数和预测因素可能需要进行更新,以达到更高的预测精度;最后,该模型还需在其他数据库或大规模的临床数据中进行验证。
综上所述,本研究不仅构建了一个具有高预测准确性的列线图,而且提供了潜在的预后生物标志物和治疗靶点,为临床决策提供可靠的理论支持。
利益冲突:所有作者均声明不存在利益冲突。
[参考文献]
[1] Siegel R L, Miller K D, Fuchs H E, et al. Cancer statistics, 2021[J]. CA Cancer J Clin, 2021, 71(1): 7–33.
[2] Lortet-Tieulent J, Ferlay J, Bray F, et al. International patterns and trends in endometrial cancer incidence, 1978—2013[J]. J Natl Cancer Inst, 2018, 110(4): 354–361.
[3] Chen W, Zheng R, Baade P D, et al. Cancer statistics in China, 2015[J]. CA Cancer J Clin, 2016, 66(2): 115–132.
[4] Sorosky J I. Endometrial cancer[J]. Obstet Gynecol, 2012, 120(2 Pt 1): 383–397.
[5] Morice P, Leary A, Creutzberg C, et al. Endometrial cancer[J]. Lancet, 2016, 387(10023): 1094–1108.
[6] Siegel R L, Miller K D, Jemal A. Cancer statistics, 2017[J]. CA Cancer J Clin, 2017, 67(1): 7–30.
[7] 张远丽, 张师前. 基于预后和分子分型的子宫内膜癌分期修订建议: 国际声音与中国现状[J]. 中国实用妇科与产科杂志, 2020, 36(3): 283–286.
[8] Bushweller J H. Targeting transcription factors in cancer-from undruggable to reality[J]. Nat Rev Cancer, 2019, 19(11): 611–624.
[9] 嚴东明, 梁健堂, 刘晓迁, 等. 胶质瘤免疫生物标志物ORMDL2在胶质瘤诊断和预后中的作用[J]. 海南医学院学报, 2023, 29(6): 428–437.
[10] Liu T, Yuan L, Zou X. Circular RNA circ-BNC2 (hsa_circ_0008732) inhibits the progression of ovarian cancer through microRNA-223-3p/FBXW7 axis[J]. J Ovarian Res, 2022, 15(1): 95–105.
[11] Lu M, Gong B, Wang Y, et al. CircBNC2 affects epithelial ovarian cancer progression through the miR-223-3p/LARP4 axis[J]. Anticancer Drugs, 2023, 34(3): 384–394.
[12] WANG Z, TANG W, YUAN J, et al. Integrated analysis of RNA-binding proteins in glioma[J]. Cancers (Basel), 2020, 12(4): 892–911.
[13] Huang H, Yang Y, Zhang W, et al. TTK regulates proliferation and apoptosis of gastric cancer cells through the Akt-mTOR pathway[J]. FEBS Open Bio, 2020, 10(8): 1542–1549.
[14] Liu Y, Zhu K, Guan X, et al. TTK is a potential therapeutic target for cisplatin-resistant ovarian cancer[J]. J Ovarian Res, 2021, 14(1): 128–138.
[15] Gao Y H, Qu S S, Cao L Q, et al. TTK predicts triple positive breast cancer prognosis and regulates tumor proliferation and invasion[J]. Neoplasma, 2022, 69(2): 274–282.
[16] Andreiuolo F, Varlet P, Tauziède-Espariat A, et al. Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: An entity with characteristic clinical, radiological, cytogenetic and histopathological features[J]. Brain Pathol, 2019, 29(2): 205–216.
[17] Qi J, Ni W. Attenuation of MAMLD1 expression suppresses the growth and migratory properties of gonadotroph pituitary adenomas[J]. Pathol Oncol Res, 2020, 26(2): 937–946.
[18] He Y, Dai X, Li S, et al. Doublecortin-like kinase 2 promotes breast cancer cell invasion and metastasis[J]. Clin Transl Oncol, 2023, 25(4): 1102–1113.
[19] Barrow T M, Wong Doo N, Milne R L, et al. Analysis of retrotransposon subfamily DNA methylation reveals novel early epigenetic changes in chronic lymphocytic leukemia[J]. Haematologica, 2021, 106(1): 98–110.
[20] Suski J M, Braun M, Strmiska V, et al. Targeting cell-cycle machinery in cancer[J]. Cancer Cell, 2021, 39(6): 759–778.
[21] Tsuda H, Hashiguchi Y, Inoue T, et al. Alteration of G2 cell cycle regulators occurs during carcinogenesis of the endometrium[J]. Oncology, 2003, 65(2): 159–166.
(收稿日期:2023–08–15)
(修回日期:2023–12–28)
