杂原子掺杂碳材料用于电合成过氧化氢的研究进展*
2024-03-01王以恒赵尔卓夏广森管少华展巨宏
王以恒 赵尔卓 夏广森 管少华 展巨宏
(1.清华苏州环境创新研究院,苏州,215163;2.北京林业大学环境科学与工程学院,北京,100083;3.清华大学环境学院,北京,100084;4.常州东方环保产业发展有限公司,常州,213025)
过氧化氢(hydrogen peroxide),即H2O2,是世界上100 种最重要的化学品之一[1],在全pH 范围内具有高氧化电位(pH 为 0 时E0=1.763 V,pH 为 14 时E0=0.878 V)[2].在合适活化条件下(如臭氧、紫外线、Fe2+等),可有效产生羟基自由基(•OH),在短时间内氧化有机污染物,将其转化成易生物降解的中间产物,因此可广泛应用于处理市政饮用水、工业废水和生活污水[2−3].H2O2的反应产物为氧气和水,安全无毒易处理,所以H2O2也被称为最清洁的氧化剂[4].目前,H2O2的工业价值接近每年40 亿美元,年需求量约550 万吨[5−6],也正因如此,探寻高效廉价制取H2O2的方法具有重要的现实意义.
蒽醌法是目前国内外生产H2O2的主要方法,世界上大约 95% 的H2O2都是通过蒽醌法生产的[7].然而,该方法无法原位生产H2O2,且合成的H2O2浓度较高,在运输过程中存在爆炸风险,储存过程中也易发生分解,运输和储存成本较高.
由于现行方法的不足,如何绿色高效地合成H2O2成为越来越多学者研究的课题.在各种方法当中,电催化two-electron oxygen reduction reaction(2e−ORR)法合成H2O2最具潜力.该方法可在常温、常压下实现H2O2的合成,相比传统的蒽醌法具有进行原位水处理、绿色安全、工艺简单、能耗较低、无污染物排放等优点,更符合当今绿色化学的发展理念.
1 氧还原反应(Oxygen reduction reaction)
1.1 ORR 反应机理
电化学氧还原反应是在一定电压下,将通入阴极的O2直接与电解液中的质子结合,实现H2O2的产生[8].ORR 是一个多步骤,多质子耦合的电子转移过程,包括多个基元步骤和反应中间产物[9−10].一般认为,ORR 反应包括两条途径,即吸附于催化剂阴极表面的O2分子通过2e−ORR 途径被还原为H2O2或4e−ORR 途径被还原为H2O[11−12](图1).

图1 2e− ORR 和4e− ORR 反应路径Fig.1 pathways of 2e− ORR and 4 e− ORR reaction
在4e−ORR 中,第一步同样是被阴极催化剂表面吸附的O2分子得到1 个电子,再从溶液中得到一个质子形成*OOH,与2e−ORR 有所不同的是,*OOH 的强吸附阻止了其从催化剂表面解吸,并最终分解成了*O 和*OH,继续得到电子和质子后形成H2O.酸性条件下4e−ORR 的化学式如(2)所示:
通过对比可以发现,O2分子和反应中间体*OOH 在催化剂阴极表面活性位点的吸附性能是发生2e−ORR 的关键.*OOH 吸附过强阻碍了其在催化剂表面的解吸,导致生成副产物H2O,加剧竞争反应;*OOH 吸附过弱则会降低反应活性,需要额外的过电位[13].ORR 反应体系中H2O2的生成是发生2e−ORR 的标志,从化学式可以看出,2e−ORR 是合成H2O2最高效的途径.因此,越来越多的研究者致力于合成兼具高2e−ORR 活性和选择性的稳定催化剂,它们具有更强O2吸附性能和合适的*OOH 结合性能,可以在最大程度上实现*OOH 的保留,进而有利于H2O2的生成.
1.2 ORR 过程的催化剂
目前,贵金属基催化剂,如Pd[14−16]、Pt[17−18]及其合金[19−20]等,它们对O2电还原具有较小的过电位和高2e−ORR 选择性.FORTUNATO[14]等利用氯化物-低金属Pd 负载的催化剂通过ORR 反应制备H2O2,发现在几何效应和电子效应的相互作用下H2O2的选择性接近100%,具有极高的质量活性[21],但是贵金属数量稀少、成本过高、易脱落造成污染的缺点限制了其规模化应用.
碳材料是常见的阴极材料,很早以前便有研究者将碳材料用于催化O2还原制备H2O2.然而,原始碳材料的表面积较小且难以提高[22],O2分子需要先溶解于溶液中再扩散到电极表面.此外,未改性的碳材料的表面活性位点也较少,这些都限制了它的催化活性.Da POZZO[23]、SALARI[24]、ÖZCAN[25]、WANG[26]等曾将原始碳材料(石墨、石墨毡、碳毡、活性炭纤维)作为催化剂用于合成H2O2,可是效果并不理想,远没有达到规模化生产的要求.因此,对碳材料催化剂阴极进行改性成为提升其电化学性能的首选方法.
根据ORR 的过程机理,催化剂改性原理主要分为两点[14,27]:
①几何效应:适当增加催化剂中活性位点之间的距离,对活性位点进行修饰,通过改变催化剂空间结构,包括催化剂活性位点分布和空间结构等,提高催化剂的催化性能.
②电子效应:根据Sabatier 的中间化合物理论,催化过程的中间产物(在本反应中为*OOH)与催化剂之间的吸附能应适中,寻找活性位点具有合适电子密度的催化剂,使其对*OOH 具有合适的吸附能.
用SPSS16.0统计软件包对实验数据进行统计学分析,3组抗剪切强度比较采用卡方检验,两两比较采用LSD-t检验;ARI记分采用Kruskal-Wallis H检验。P<0.05为差异有统计学意义。
当前,有关电催化2e−ORR 合成H2O2过程催化剂的改性重点集中在向碳基基底引入其他碳材料(炭黑[28]、乙炔黑[29]、石墨烯[30]等)、杂原子(N[31−34]、O[35−37]、F[38−39]等)掺杂、材料结构的调控、反应条件的优化等方法提高O2分子在还原过程中与催化剂表面结合的活性、选择性以及催化剂的稳定性.由于掺入的杂原子具有不同的电负性,碳基体的局部电荷和自旋密度发生了显著改变,促进了对O2的吸附,降低了反应能垒,增强了碳材料电催化ORR 的活性[40−41].杂原子掺杂后的碳材料与贵金属基催化剂和原始碳材料相比具有可操作性好、电化学性能优异、成本低廉等优点,可用于原位处理市政饮用水、工业废水和生活污水,拥有良好的发展前景(图2).表1 展示了部分杂原子掺杂碳材料催化剂电合成H2O2的性能.
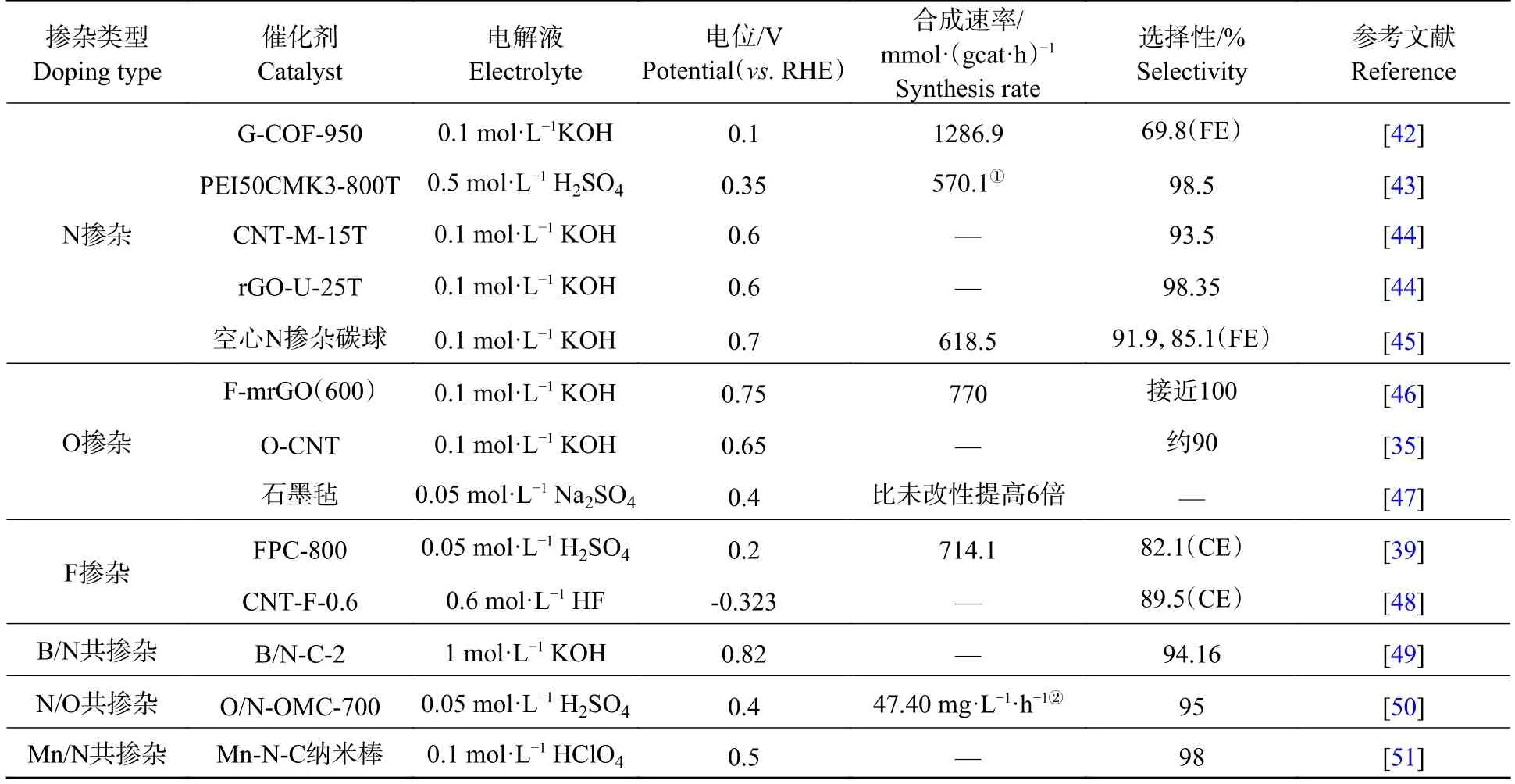
表1 部分杂原子掺杂碳材料催化剂电合成H2O2 的性能Table 1 Performance of partially heteroatom-doped carbon material catalysts for the electro-synthesis of H2O2

图2 杂原子掺杂碳材料改性优点Fig.2 The advantages of heteroatom-doping carbon material modification
2 杂原子掺杂碳材料(Heteroatom-doped carbon materials)
2.1 N 掺杂
N 掺杂碳材料是指将N 原子通过化学键连接或结合到碳骨架中产生的新型材料.N 原子和C 原子尺寸相接近,在N 原子取代C 原子的过程当中,碳材料骨架所收到的破坏程度较小,具有较高的稳定性和耐久性.此外,N 可以改变相邻C 原子上的电荷分布,从而产生更多的活性位点,促进ORR 的发生[52].N 原子掺杂碳材料中的N 主要有4 种类型:石墨-N、吡啶-N、吡咯-N 和氧化态-N(图3),由于自身电子结构差异,不同N 物种具有不同的结构性能.例如,由于吡啶-N 物种具有未杂化的孤对电子,有较强的给电子能力,可以有效提供 Lewis 碱位点,所以富含吡啶-N 物种的N 掺杂碳材料催化剂在ORR 中具有更优异的催化性能[53].
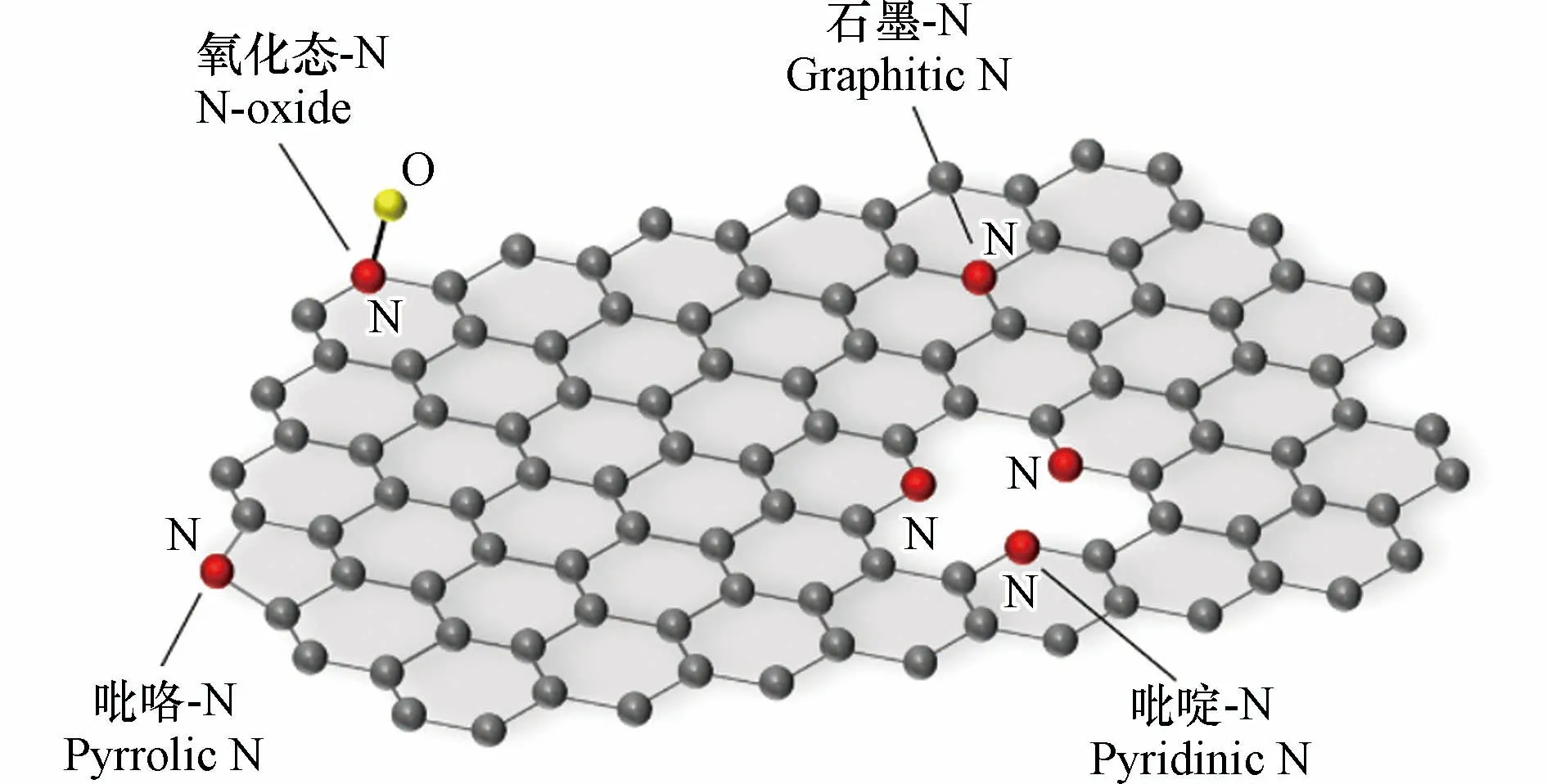
图3 N 掺杂碳材料中N 的四种类型示意图Fig.3 Four types of nitrogen in N-doped carbon materials
目前的N 掺杂方法尚不能建立有效的结构性能关系,所以明确不同键合类型的N 掺杂效果,识别N 掺杂碳材料的活性位点具有重要意义.2012年,FELLINGER 等[31]采用离子液体N-丁基-3-甲基吡啶二氰胺(BMP-dca)作为前体合成N 掺杂介孔碳,催化剂产生1 g H2O2的实际比能量为0.241 W·(gcat·h)−1,法拉第电流效率达65.15%.N 掺杂介孔碳催化剂有着较高的2e−ORR 选择性,这是因为掺杂到碳材料中具有较高电负性的N 原子可以通过破坏π 共轭系统完整性并诱导电荷再分布来激活π 电子,从而改变碳材料的吸附性能,有利于H2O2的生成.
2020年,ZHANG 等[42]采用共价有机框架(COF)作为前体,在Ar 气氛下热解,加热至恒温合成了N 掺杂催化剂 G-COF-T(G-COF-550、G-COF-750、G-COF-950).X 射线光电子能谱(XPS)分析发现催化剂中总氮含量随热解温度的升高而降低:吡啶-N 和吡咯-N 的含量减少,石墨-N 的含量增加.其中,950 ℃下处理的G-COF 中石墨N 的含量从0.51%增加到1.86%,占总氮的比从13.5%增加到60.7%(图4a).在0.1 mol·L−1的KOH 溶液中,该催化剂H2O2生成速率为1286.9 mmol·(gcat·h)−1,法拉第效率达69.8%.进一步推断认为,石墨-N 可以通过给p-π 共轭体系提供一个单电子来诱导电荷再分配,从而增强氧气在相邻的C 原子上的吸附,将其转变为中间体HOO*后,石墨-N 的不稳定结构趋于电荷平衡,促使*OOH 解吸(图4b).因此,高密度、高活性的石墨-N 有助于N 掺杂的碳材料高效催化2e−ORR 的发生.
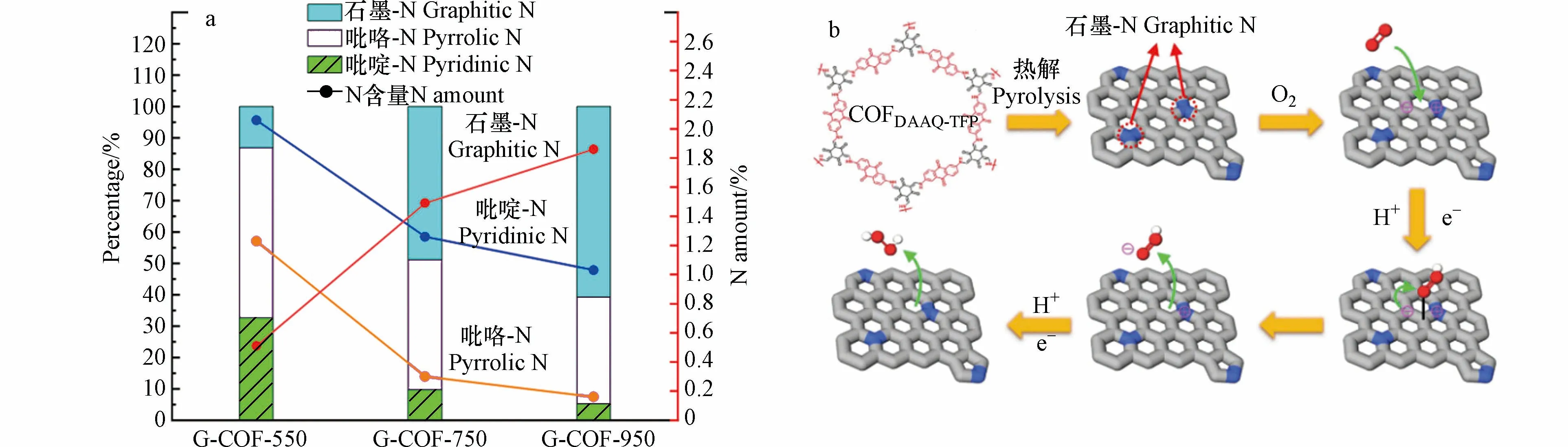
图4 (a)G-COF 在不同温度处理后不同N 掺杂类型的比例及含量;(b)N 原子掺杂碳材料的可能机理[42](红色:氧原子;白色:氢原子;灰色:碳原子;蓝色:氮原子).Fig.4 (a)Percentages and amount of nitrogen functionality present in catalysts ;(b)Proposed mechanism of electrochemical 2e− ORR on COF-derived N-doped carbon catalysts(Red: oxygen;white: hydrogen;grey: carbon;blue: nitrogen)[42].
此外,SUN 等[43]将有序介孔碳(CMK-3)与聚乙烯亚胺(PEI)直接热解制备得到N 掺杂多孔碳材料PEI50CMK3-800T.分析发现,在0.05 mol·L−1H2SO4溶液,电位为0.35 V(vs.RHE)的条件下,H2O2的选择性从未改性时的77.2%大幅提高到98.5%,;在中性条件下,H2O2的生成速率达到570.1 mmol·(gcat·h)−1.XPS 结果表明,在不同pH 条件下,不同类型的N 掺杂效果不同.酸性条件下,吡啶-N 在催化过程中起关键作用,而在中性和碱性条件下,石墨-N 基团的催化活性更高(图5).最近有研究表明[54−56],ORR 过程中吸附的*OOH 中间体可以改变催化剂的催化活性位点或结构基序的直接化学环境.在酸性条件下,—OH 与相邻吡啶-N 的碳原子共价结合,形成一个羟基吡啶-N 结构,该结构中N 的1s 结合能由初始的398.8 eV 上升到400.2 eV,与吡咯-N 的结合能非常接近.与之类似的是,—OH 与相邻石墨N 的碳原子也会共价结合,使N 的1s 结合能降低至400 eV 左右,同样与吡咯-N 的结合能接近.吡咯-N 含量的增加证实了其作为有效活性位点结构促进了2e−ORR 的发生.
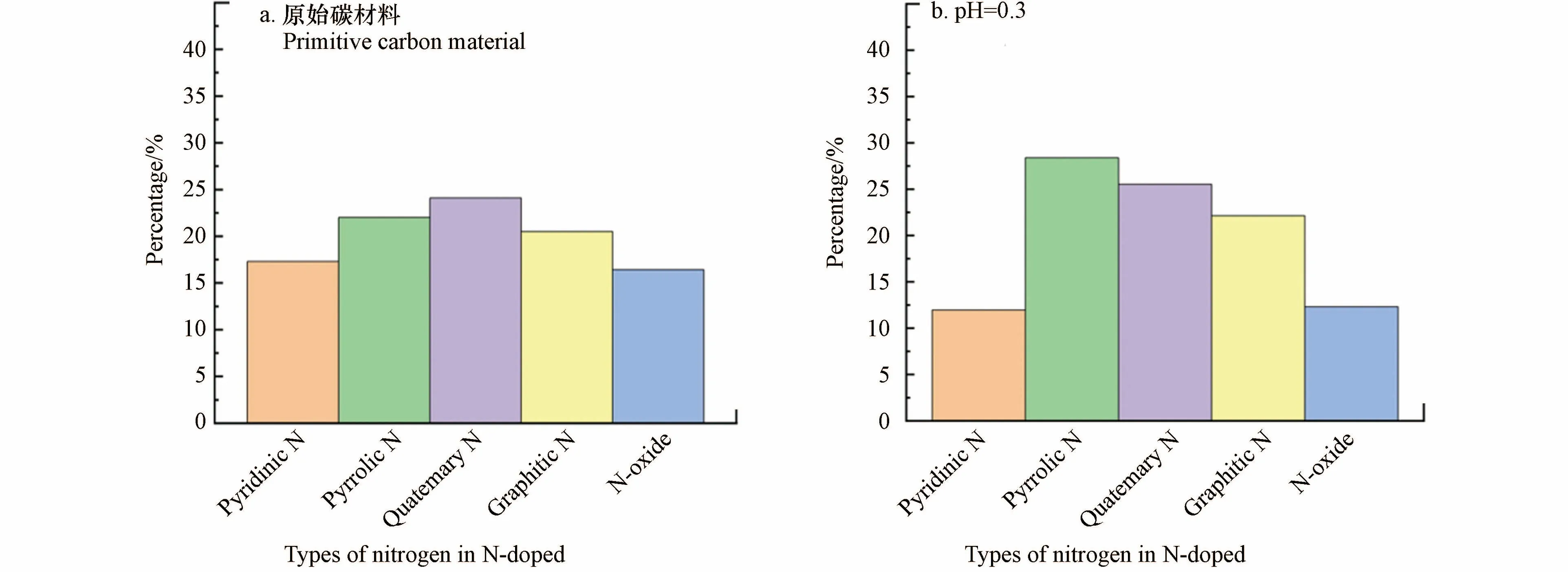
图5 催化前(a)和催化后PEI50CMK3-800T 催化剂中不同N 掺杂类型的在不同pH 条件下((b)pH 0.3、(c)pH 7 和(d)pH 13)相对含量的变化[43].Fig.5 (a—d)The relative content change of various nitrogen species of the PEI50CMK3_800T catalyst before(a)and after the catalysis in(b)pH 0.3,(c)pH 7,and(d)pH 13[43].
2022年,WAN 等[44]通过微波辅助脉冲加热(MAPH)方法,成功制备了具有多种键合类型(石墨-N,吡咯-N 和吡啶-N)的N 掺杂单壁碳纳米管(CNT-D-40T、CNT-U-15T、CNT-M-20T)(图6a).N 掺杂可以通过扰乱π 共轭体系的完整性来激活C 的π 电子的ORR 活性.π 电子的活性与其电子亲和力密切相关,在吡啶-N 构型中的N 原子会与两个碳原子结合,为π 体系贡献一个p 电子,而吡咯-N 可以为构型中的π 共轭体系贡献两个p 电子.除此以外,计算电子转移数发现,吡咯-N 掺杂的CNT-U-15T 在很宽的电位范围内(0.30—0.65 V,vs.RHE)具有最小的电子转移数值,与2 最为接近,小于石墨-N 掺杂的CNT-D-40T 和吡啶-N 掺杂的CNT-M-20T(图6b).这些都无疑证明了吡咯-N 掺杂的优异2e−ORR 催化性能.
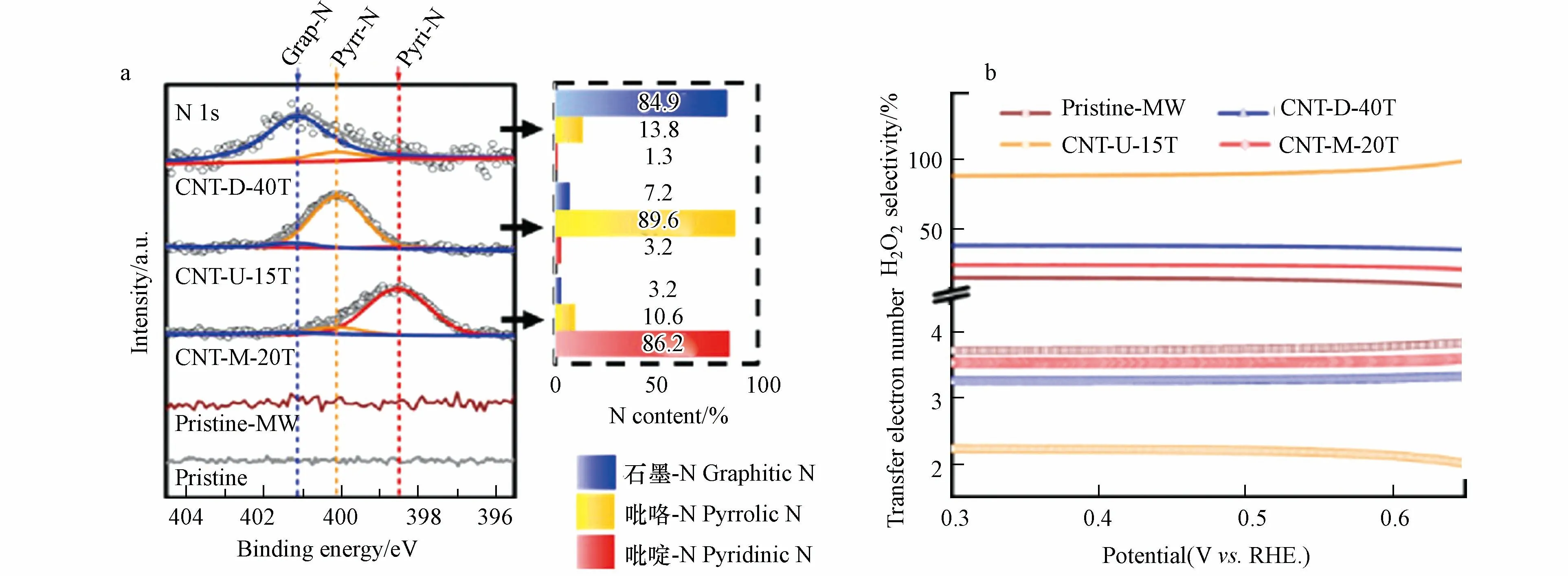
图6 (a)CNT-D-40T、CNT-U-15T、CNT-M-20T 中石墨-N,吡咯-N、吡啶-N 的含量;(b)电位扫描过程中H2O2 生成的选择性和电子转移数(n)[44].Fig.6 (a)Graphite-N,pyrrole-n and pyridine N contents in CNT-D-40T,CNT-U-15T and CNT-M-20T;(b)H2O2 selectivity and electron transfer number(n)during the potential sweep [44].
近期,HU 等[45]将聚多巴胺碳球碳化,并在Ar 气氛下进行进一步高温处理得到了具有丰富微孔的空心N 掺杂碳球,其在0.1 mol·L−1KOH 溶液,0.7 V(vs.RHE)的条件下,相比于原始碳材料(77.0%和61.8%),具有更高的2e−ORR 选择性[91.9%,0.7 V(vs.RHE)]和法拉第效率[85.1%,0.7 V(vs.RHE)],H2O2生成速率达618.5 mmol·(gcat·h)−1.研究发现,N 掺杂碳球中更多的微孔体积通常有利于活性位点的暴露,此外,微孔隙率还可以减少H2O2的停留产物,从而防止进一步还原成为H2O,这也证明了空心结构的杂原子掺杂碳材料极大地有利于2e−ORR 合成H2O2.
2.2 O 掺杂
O 掺杂可以将各种含氧官能团(OG)引入碳材料表面,例如羧基(―COOH)、醛基(―CHO)、羰基(—C=O)、醚键(C—O—C)等.O 掺杂的碳材料具有合成简单、成本低廉的特点,引入的OG 会显著地改变其表面性质,如反应性和亲水性.与N 掺杂类似,O 掺杂也可以显著改变相邻C 原子上的电荷分布,从而调整*OOH 在碳材料表面的吸附特性,促进2e−ORR 的发生.因此,氧化碳纳米管(O-CNT)[35]、氧化石墨烯[46]、活性炭黑[57]等O 掺杂碳材料作为2e−ORR 催化剂的研究已经取得一定进展.
2010年,MATSUBARA 等[47]就利用120 °C 的2 mol·L−1硫酸和1:1 浓硝酸处理多壁碳纳米管(MWCNTs),发现了在表面形成的OG 可以使反应过电位降低.2013年,Zhou 等[36]通过阳极氧化石墨毡发现,酸性的OG 可以提高碳材料表面的亲水性能,更易于获得水中的溶解氧,促进电极和电解质之间的电子传输.2014年,MIAO 等[37]在硫酸中电化学氧化石墨毡电极,将某些OG 经过修饰后引入石墨毡电极表面,发现处理后的石墨毡电极阴极上2e−ORR 反应得到增强,H2O2合成效率提高到了原来的6 倍,进一步证明了OG 对ORR 反应的促进作用.
关于OG 提高碳材料催化活性的机理,由于不同实验室采用不同的碳材料和氧化过程,给出的解释也不尽相同.2018年,LU 等[35]通过浓硝酸氧化的方式成功制备了O-CNT.与标准的CNT 相比,OCNT 在0.1 mol·L−1KOH 溶液,0.2 mA 的电流下的过电位降低了约130 mV,将2e−ORR 反应选择性从60%提高到了90%.与此同时,LU 等发现O-CNT 的活性和选择性都与其氧含量呈正相关,不同结构的催化活性由反应中间体与催化剂活性中心的结合能决定(图7a).LU 等通过进一步密度泛函理论(DFT)计算,根据*OOH 吸附能的变化,绘制了不同OG 通过2e−ORR 合成H2O2的火山图(图7b),结果表明,在石墨烯基面和边缘上的醚键具有极高的2e−ORR 反应活性,它们的过电位分别为0.02 V 和0.06 V,这与贵金属催化剂Pt-Hg 和Pt-Au 相当[20].
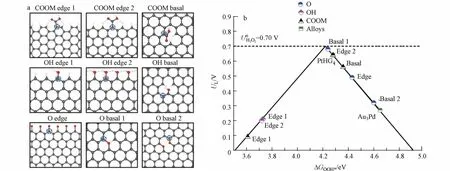
图7 (a)不同官能团的ORR 活性,用蓝色圆圈表示的碳原子为活性中心[44];(b)不同OG 根据*OOH 吸附能合成H2O2 的活性火山图[44],包括了Pt–Hg 和Pd–Au 合金的活性[20].Fig.7 (a)Different oxygen functional group type configurations examined in this study.The carbon atoms denoted by a blue circle are the active sites under investigation [44];(b)Calculated two-electron(solid black)ORR-related volcano plot for the electro-reduction of oxygen to H2O2 displayed with the limiting potential plotted as a function ofΔ GOOH*[44],including the activity of Pt --Hg and Pd --Au alloys[20].
LU 等在利用浓硝酸氧化制备O-CNT 的实验中,未提到是否会引入N.对此,一些研究者提供了其他的氧化方法.Kim 等[46]利用氧化石墨烯材料,通过热还原富集氧化石墨烯中的醚键和环氧树脂,使用碳纸成功合成了多层mrGO 催化剂(F-mrGO(600)),该催化剂过电位小于10 mV 且具有极高的2e−ORR 活性和稳定性,选择性更是接近100%.此外,WANG[57]等通过在500 ℃空气气氛中退火来提高活性炭黑(aCB)中的C—O 键和O—H 键浓度,有效将过电位降到了5 mV 以下,在0.75V(vs.RHE)下H2O2的最大生成速率达770 mmol·(gcat·h)−1.这种简单的活化方法可以在不改变含氧量的情况下操纵含氧基团的比例,从而准确地将催化活性与含氧基团相关联(图8a).根据实验结果和DFT 计算,绘制出火山图,得出添加醚键和醛基的碳材料催化剂表面活性位点对*OOH 的吸附能最低,过电位较小,是生成H2O2的活性中心(图8b).
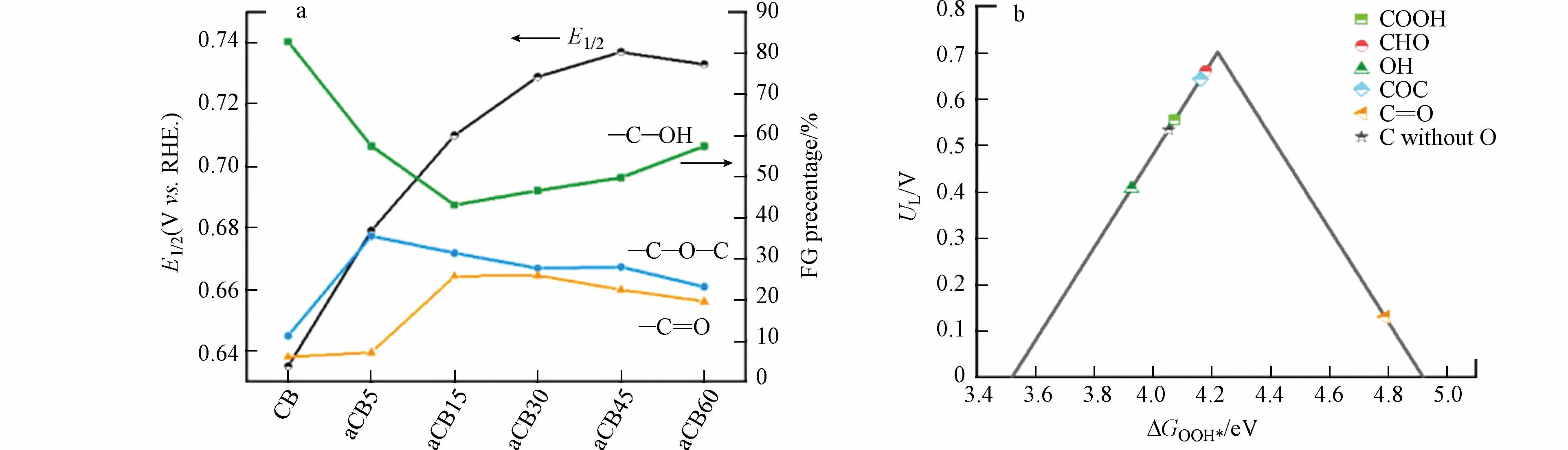
图8 (a)不同退火时间的催化剂中不同OG 的催化活性和比率;(b)不同OG 根据*OOH 吸附能合成H2O2 的活性火山图[57].Fig.8 (a)Catalytic activity and ratio of the different oxygen-containing functional groups as a function of the series catalyst annealed with different activation time;(b)ORR activity volcano plot of the different oxygen-containing functional groups modified carbon for the synthesis of H2O2 according to the adsorption energy of HOO*[57].
先前研究表明,电催化剂上各位点吸附反应物的能力与其活性直接相关[58].杂原子掺杂后电荷密度和自旋密度的再分布会影响碳材料中活性位点的分布,在掺杂剂附近具有高电荷密度或高自旋密度的碳原子可能成为活性位点[59].2022年,XIE 等[60]基于DFT 计算,引入福井函数,用以识别活性位点并系统地研究O 掺杂碳材料(OCM)的活性.结果表明,醚键和羰基可能是2e−ORR 的高活性来源.通过研究不同OG 共掺杂的碳材料的2e−ORR 活性,XIE 等发现不同OG 之间具有显著的协同效应.OCM 中所有碳原子*OOH 吸附游离能的计算表明,除醌外的所有OG 的引入都能增加碳材料中2e−ORR 活性位点的数量(图9a).醚键和羰基分别可以引入9 个和7 个活性位点,相较于其余OG 可以更大程度地提高OCM 的催化活性.XIE 等还绘制的自由能变化图总结了掺杂各种OG 对碳材料催化活性的影响,当掺杂羰基或醚键时,自由能变化相对平稳,过电位分别为0.05 V 或0.15 V,而当掺杂其他OG 时,碳材料的自由能图非常陡峭,过电势至少为0.5 V(图9b),这意味着低过电位的材料会具有更多的活性位点.最终得到不同OG 的催化活性大小如下:醚键>羰基>>羟甲基>羟基≈环氧≈羧基≈醛>内酯≈吡喃>碳≈醌.这也与Chen[61]等得出的结论:羰基>羧基≈羟基一致.

图9 (a)活性位点的数量和(b)不同含氧官能团在H2O2 合成过程中的自由能变化图[60]Fig.9 (a)The number of active sites and(b)the free energy diagram in different O-doped carbon models [60]
2.3 F 掺杂
F 作为第二周期卤族元素,具有极高的电负性,可以通过诱导相邻的碳极化以产生活性位点,从而增强*OOH 在催化剂表面的特异性吸附.2011年,Sheng 等[62]以乙炔黑粉(ABP)为催化剂,聚四氟乙烯(PTFE)为粘结剂,制备了一种新型的乙炔黑-聚四氟乙烯电极,在pH=3,电流密度为 20 mA·cm−2的条件下,H2O2的平均生成速率达到58.9 mg·L−1·h−1·cm−2,电流效率效率高达92.7%,展示出了含F 碳材料良好的ORR 反应特性.
先前有研究表明,F 掺杂碳材料主要通过抑制4e−ORR 反应来提高2e−ORR 合成 H2O2的活性和选择性[48].为了进一步讨论不同密度和分布的F 掺杂的碳材料对H2O2电合成的活性和选择性的影响,ZHAO 等[39]采用铝基MOF(MIL-53(Al))作为前体在不同碳化温度下合成了F 掺杂的分级多孔碳(FPC-700、FPC-800、FPC-900、FPC-1000).将FPC 与未掺杂F 的碳材料(HPCs)进行比较,发现 FPC 在ORR 反应中具有更高的电流密度,更高的负起始电位,和更低的电荷转移电阻(图10a、b),FPC-800 270 mV 的过电位,也低于HPC-800 的311 mV.这些观察结果表明,将F 原子掺入碳框架中可以最大限度地降低电荷转移阻力,提高催化剂活性,加速ORR 反应(图10c).FPC-800 在pH=1,外加电位为0.2V 条件下的H2O2生成速率达到714.1 mmol·(gcat·h)−1,电流效率达82.1%,表现出出色的2e−ORR催化性能.
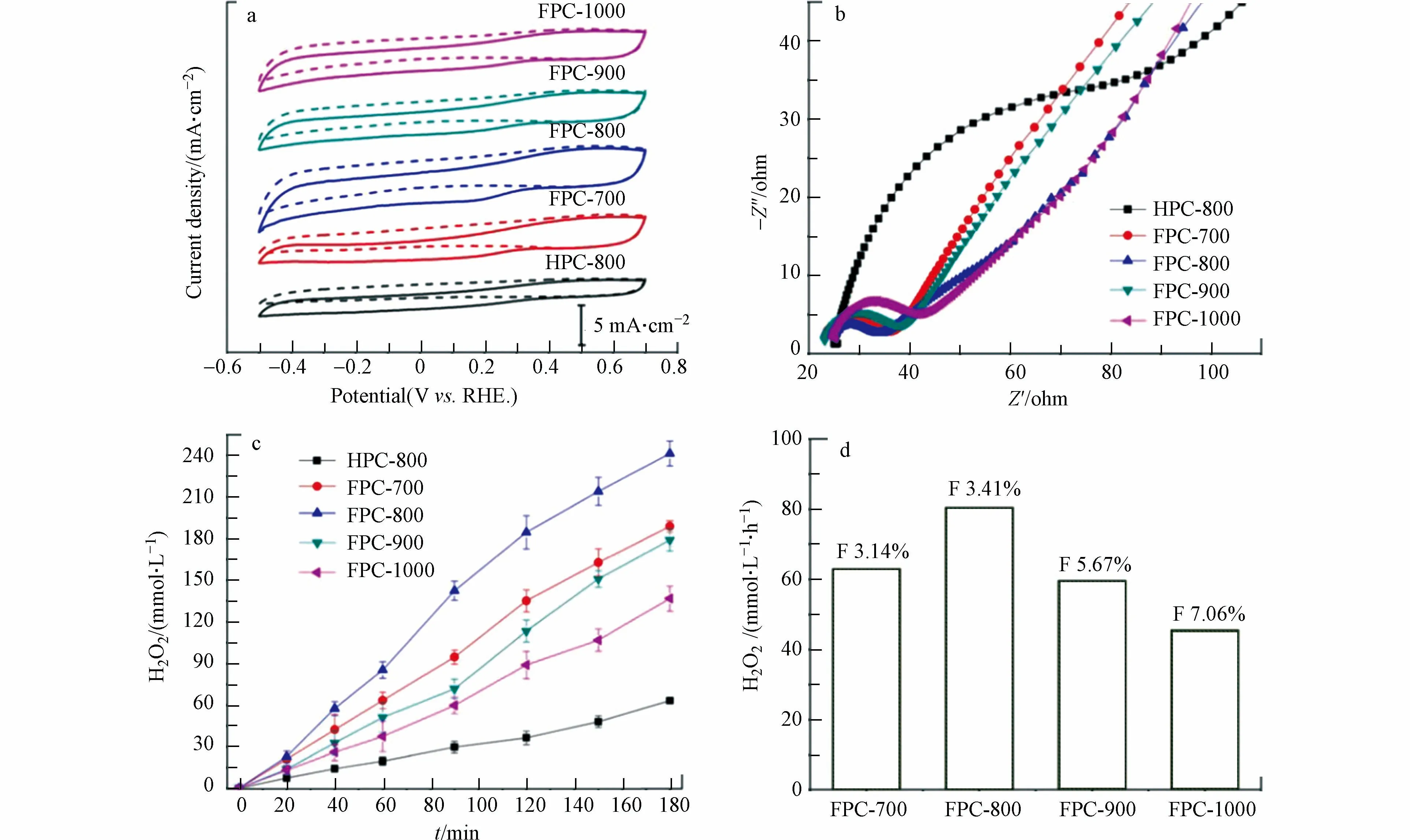
图10 (a)FPCs 和HPC-800 在(虚线-Ar)和(实线-O2)中的循环伏安曲线(扫描速度10 mV·s−1,0.05 mol·L−1 H2SO4);(b)FPCs 和HPC-800 在0.05 mol·L−1H2SO4 中的EIS 数据(交流电压为5 mV,频率范围在100 kHz—0.1 Hz);(c)FPCs 和HPC-800 的H2O2 产量随时间的变化;(d)FPCs 在不同F 掺杂含量下H2O2 的生成速率[39].Fig.10 (a)CV curves of FPCs and HPC-800 in(Ar-dash line)or(O2-saturated solid line)0.05 mol·L−1 H2SO4 solution with a scan rate of 10 mV·s−1;(b)EIS data of FPCs and HPC-800 in O2-saturated 0.05 mol·L−1 H2SO4 solution with an AC of 5 mV and a frequency range between 100 kHz and 0.1 Hz;(c)The concentration of H2O2 produced by FPCs and HPC-800 obtained at different carbonization temperature;(d)H2O2 production ration with F content [39]
随着热解温度在700—1000°C 范围内变化,F 的掺杂含量从3.10%增加到7.06%(图10d).一般认为,在碳材料中掺杂F 原子会破坏碳的电荷均匀性,并为H2O2的产生提供活性位点.然而,当材料中F 原子含量过高时,材料的石墨化程度会随着F 原子含量的提高而降低,从而导致电子电导率降低,产生 H2O2的活性降低.
对此,ZHAO 等构建了3 种不同的F 掺杂碳材料(Gr-F、Gr-F2和Gr-F3)以及未进行F 掺杂的纯石墨碳(Gr)模型(图11),确定了反应中间体在不同类型F 掺杂碳材料和纯石墨碳表面的吸附能差异.对比发现,Gr-F、Gr-F2和Gr-F3相比Gr 对O2分子具有较弱的吸附能,这有利于O2分子在催化剂表面的活化;Gr-F2具有较弱的OOH 结合能,可以促进H2O2的生成.因此,Gr-F2被认为是最有利于2e−ORR 的F 掺杂类型.
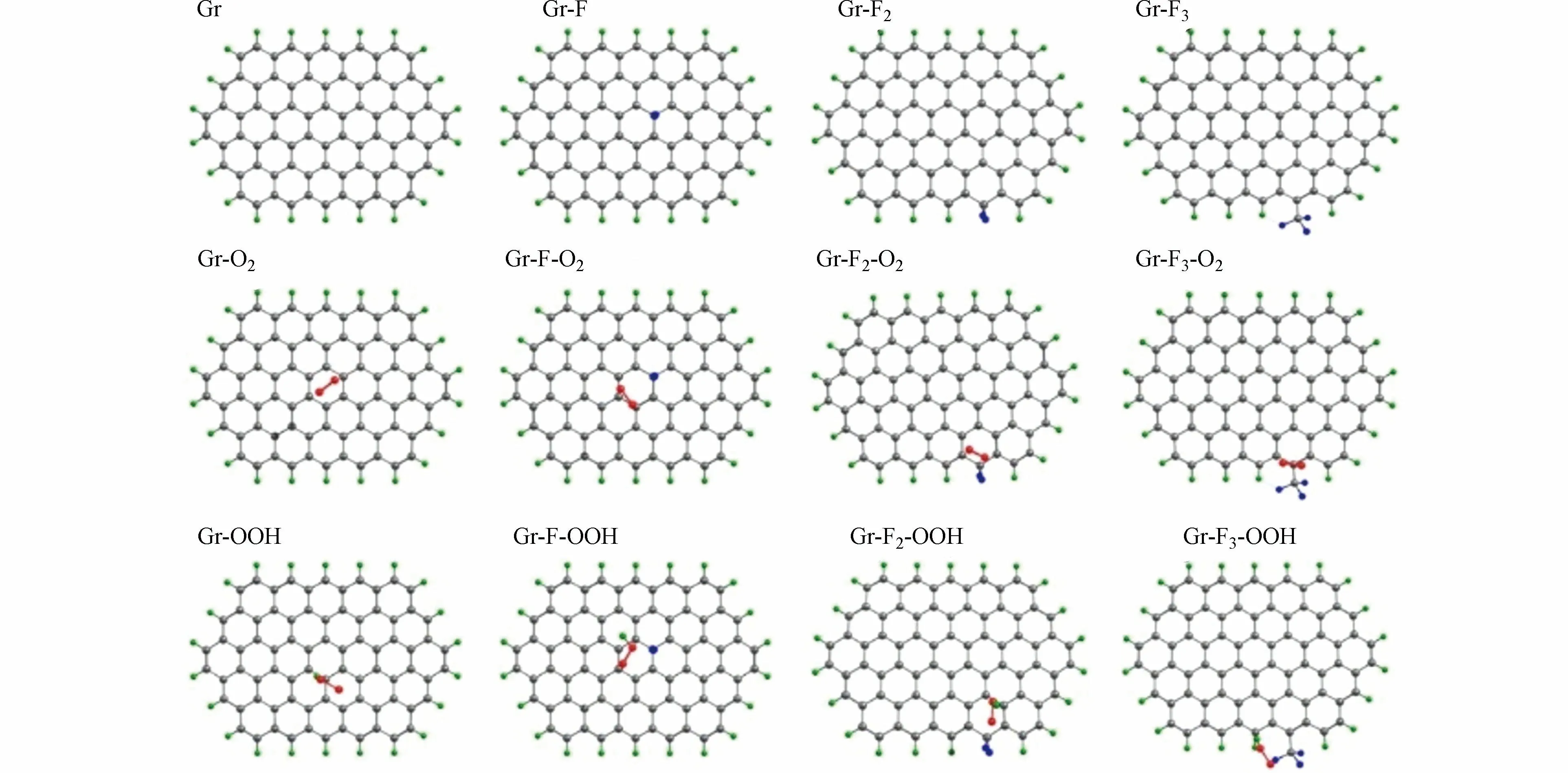
图11 不同类型的F 掺杂碳材料和未改性碳材料对O2 和OOH 的吸附计算模型[39]Fig.11 The computational models for adsorption of O2 and OOH on different types of F-doping and pure carbon materials[36]
WANG[63]等采用F 改性CNT,以F 掺杂碳纳米管(F-CNT)制备的气体扩散电极(GDE)作为阴极生产H2O2.实验发现,用0.6 mol·L−1HF 溶液制备的F-CNT(CNT-F-0.6)具有更高的电产H2O2浓度和电流效率,分别达到47.6 mg·L−1和89.4%,高于未改性时的的27.6 mg·L−1和70.1%.与Zhao 等的结论一致:WANG 等也认为F 原子掺杂可以破坏碳的电荷均匀性,促进O2的吸附和*OOH 的形成,而CF2与*OOH 之间较弱的吸附能又有利于H2O2的形成.
2.4 多原子掺杂
由于不同掺杂原子之间存在的协同效应,双杂原子或多杂原子掺杂往往会更大程度地增加催化剂表面的活性位点和2e−ORR 选择性,从而起到更好的改性效果.基于同族元素化学性质相似的特点,近年来出现了许多不同杂原子组合掺杂改性碳材料以合成H2O2的实验,例如B/N[49,64]、N/O[50]、N/S[65−66]、O/F[67]等,它们具有优秀的2e−ORR 选择性和较高的H2O2产率,进一步验证了杂原子掺杂改性碳材料具有广阔的应用前景.
2021年,LI 等[49]通过将聚乙烯醇-石墨烯、硼砂和聚苯胺凝胶化后进行热解,成功制备具有可调B、N 构型的B/N 共掺杂多孔碳气凝胶.H2O2的合成选择性在碱性溶液,在外加正电位(0.6 Vvs.RHE)的条件下达到了94.16%,表现出优异的电催化性能.由于B(2.0)与C(2.5)、N(3.0)原子电负性的差异,使B 原子上的电子逐渐向C、N 原子积累,这有利于B 原子产生空轨道以接受来自HOO*中间体的电子.DFT 研究发现,hBN-N6-B1 位点具有最低的过电位和最小的自由能势垒(图12a、b),hBN 与邻近的吡咯-N 位点相结合是进行2e−ORR 合成H2O2最活跃的位点(图12c).与LI 等[49]的想法类似,LIU 等[64]通过水热和热解工艺设计合成了BCN 催化剂,B 原子能有效结合N 原子,使两种原子进入石墨晶格,取代C 原子形成BCN,其在较宽电位范围内的2e−ORR 选择性超过70%,可稳定工作12 h.
鉴于N、O 原子单独掺杂时均取得了较为良好的效果,QIN 等[50]利用己胺(HMT)、聚环氧乙-b-聚环氧丙-b-聚氧化乙二醇三嵌段共聚物(Pluronic F127)和间苯二酚通过高压水热和碳化处理成功制备了N/O 共掺杂有序介孔碳材料,其2e−ORR 选择性高达95%,在pH=3,外加电位(-0.2 Vvs.RHE)的条件下,H2O2合成速率达47.40 mg·L−1·h−1.研究发现,吡啶N 与C—O—C/COOH 的结合相比于其他结构大大降低了OOH*的吸附能(图13),从而促进了2e−ORR 反应.

图13 *OOH 在12 个碳模型上的吸附能Fig.13 *OOH absorption energy on 12 configurations
HUANG[68]等以Co—N—C 催化剂(ZIF-67)和Ti3C2Tx为前体物,合成了过渡金属碳氮共掺杂碳材料催化剂,在酸性条件下H2O2合成效率达94.1 mmol·L−1·h−1.光谱分析发现,与ZIF-67 相比(13.32%),掺杂Ti 的催化剂中Co 的含量有所降低(4.78%)(图14a、d),这是由于Ti3C2Tx的层状结构,有利于Co—N—C 的均匀分布和去除暴露的Co 纳米颗粒[69].此外,Co—N—C/Ti3C2Tx的自旋轨道分裂值(ΔCo=14.4 eV)比Co—N—C(ΔCo=13.1 eV)大,这表明Co 原子的电荷密度状态较低,使其具有合适的*OOH 结合能,以促进H2O2的形成.此外,在Co—N—C/Ti3C2Tx中,吡啶-N 占总配位N 的主导地位(39.56%),高于Co—N—C(31.86%)(图14b、e),C—O—C 比例(8.30%)也高于Co—N—C(1.02%)(图14c、f).C—O—C 具有优异的2e−ORR 催化活性,XPS 结果表明,Ti3C2Tx上的氧官能团可以有效地微调Co 原子的电子结构,使其处于较低的电荷密度状态,这将使*OOH 具有合适的结合能,从而实现高效电合成H2O2.
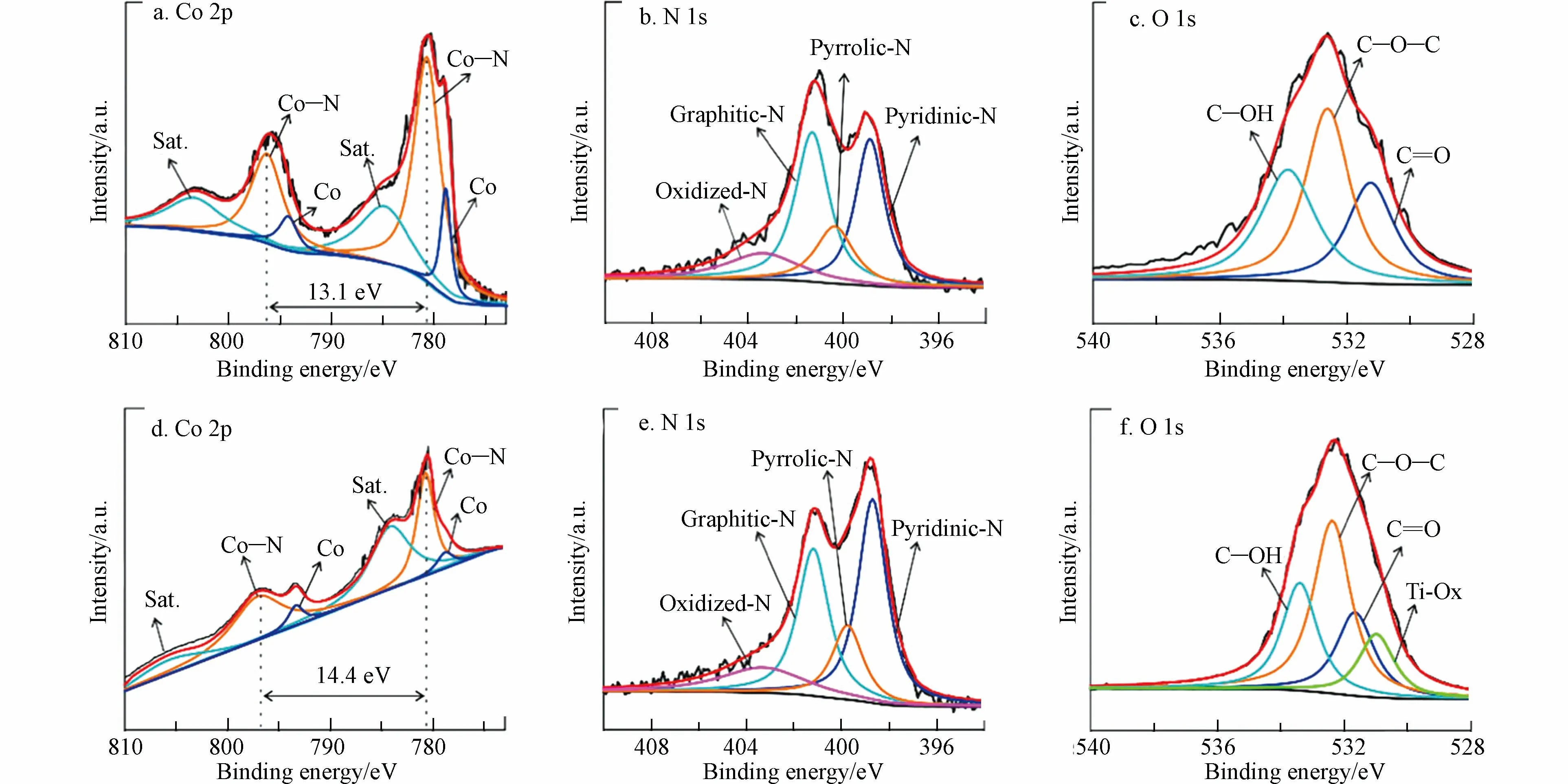
图14 (a,d)Co—N—C 和 Co—N—C/Ti3C2Tx 的 Co 2p 光谱;(b,e)Co—N—C 和 Co—N—C/ Ti3C2Tx 的 N 1s 光谱;(c,f)Co—N—C 和 Co—N—C/Ti3C2Tx 的 O 1s 光谱[68].Fig.14 (a,d)Co 2p spectra of Co—N—C and Co—N—C/Ti3C2Tx.(b,e)N 1s spectra of Co—N—C and Co—N—C/Ti3C2Tx;(c,f)O 1s spectra of Co—N—C and Co—N—C/Ti3C2Tx[68].
除此以外,其他不同类型的杂原子掺杂组合也展现出了良好的应用潜力.2016年PERAZZOLO 等[66]利用二氧化硅作为无机模板,将其和有机前体物(1,10-菲罗啉、吩噻嗪、二苯并噻吩和)溶解与丙酮中,加热后冷却制成了N/S 共掺杂介孔碳.其在氧化还原反应中显示出高达80%的2e−ORR 选择性,作为电解池阴极时可更快更有效地降解甲基橙,矿化了53%的TOC.后来,ZHANG 等[70]采用5-氨基-2-巯基-1,3,4-噻二唑(AMT)作为N 和S 掺杂剂,通过简单的热退火工艺制备了无金属的“石墨烯合金”(NSrGO).电化学阻抗谱(EIS)和循环伏安法(CV)结果显示,N 和S 共掺杂产生的协同效应引入了不成对电子,提供的活性位点增强了H2O2的吸附能力,并将电荷转移电阻从原始的360.1 Ω 降低到了17.7 Ω,增强了碳基质的导电性,进一步提高了电子转移效率.2019年,JASPER 等[70]将氮基三乙酸锰前驱体水热合成后热解和酸洗,合成了Mn—N—C 纳米棒.使用旋转环盘电极评估其2e−ORR 活性和H2O2选择性发现,Mn—N—C 纳米棒不仅在酸性介质中表现出高活性,而且在外加电位(0.5 Vvs.RHE)时表现出的H2O2选择性高达98%.随后使用Mn—N—C 纳米棒催化降解亚甲基蓝(MB),将工作电极浸入电解质中以排除吸附影响,通电后观察到MB 浓度逐渐明显下降,证实了电催化亚甲基蓝分解的发生.近期,GU 等[67]将MWCNTs 分散在H2SO4与HF 的混合溶液中,干燥退火后浸泡于聚四氟乙烯合成了新型O/F 共掺杂石墨毡气体扩散阴极(GF-GDC),在pH=7,20 mA·cm−2的电流密度下1 h的H2O2累计浓度高达232.2 mg·L−1,电流效率高达83%,且15 个周期内电流效率仅降低10%,具有良好的稳定性.
3 总结与展望(Summary and Outlook)
相比于国内外目前广泛采用的蒽醌法,电催化2e−ORR 是一种有效、绿色、安全制备H2O2的方法,具有良好的应用前景.不同类型的杂原子掺杂的碳材料可以不同程度地提高碳材料催化剂的活性、2e−ORR 选择性、循环稳定性等,是催化ORR 合成H2O2的最佳方法之一.杂原子掺杂碳材料催化能力的提高与掺杂原子类型和形成的掺杂结构密切相关,在众多科研工作者的努力下,不同杂原子的作用机理已经逐步明晰.其中,N 掺杂中形成的吡咯-N、O 掺杂中形成的醚键和F 掺杂中形成的CF2具有优秀的催化活性,以及多原子掺杂中不同原子之间形成的协同效应,均可以改善催化剂表面活性位点的分布,极大提高材料对O2分子的吸附能力以及对ORR 反应中间体*OOH 的脱附能力,发挥最佳的电化学催化性能.
与此同时,杂原子掺杂碳材料催化剂还不可避免地存在着一些问题,①不同实验中制备的碳材料催化剂结构复杂,重复性差,理论与实际难以形成一致的结论,增加了探明催化位点对O2作用机理的难度;②催化剂的性能需要进一步提高以满足实际应用;③催化剂的制备方法还需进一步简化,以满足工业领域的大规模生产.④多杂原子掺杂时对ORR 选择性的提升方向具有明显的差异,需进一步加以鉴别.随着科学技术的不断进步,先进的实验方法和计算手段的加入,杂原子掺杂碳材料催化ORR 的反应机理会变得更加明晰,2e−ORR 的催化活性会得到进一步提高,合成后的H2O2可应用于分散式水处理场景,极大地提高污水处理效率.随着技术和工艺的不断升级,H2O2的处理能力会不断增强,应用领域将不断扩大,这也势必会推动水处理技术的革新与进步.
