Integrative analysis of transcriptomic profile reveals potential roles of miRNAs in regulating development of Marsupenaeus japonicas*
2024-02-27JingWANGLongjunPUXiaojuanZHANGCuicuiLIANGDandanDONGJiantaoGUANHuarongGUO
Jing WANG, Longjun PU, Xiaojuan ZHANG, Cuicui LIANG, Dandan DONG,Jiantao GUAN, Huarong GUO,2,**
1 Ministry of Education Key Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China
2 Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao 266003, China
3 Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, China
Abstract Regulation of microRNAs (miRNAs) on various biological processes has been a surprising and exciting field.Identification of miRNAs is the first step to comprehensively understand their functions.However, attempts on global identification and functional verification of miRNAs are very limited in penaeid shrimp Marsupenaeus japonicus, an economically important aquatic species.By performing an integrated analysis of transcriptomic profile from gastrula embryos of M.japonicus, 21 conserved miRNAs in M.japonicas (mja-miRNAs), belonging to 19 miRNA families, were identified and characterized.Of the 21 mja-miRNAs, 15 miRNAs were successfully verified to be predominantly expressed in gastrula stage, where they displayed dynamic expression patterns compared with those in naupliuin stage.Based on perfect or near-perfect match to target region, 120 target genes were predicted at transcriptome-wide level.Noteworthy, gene ontology (GO) classification and metabolic pathway annotation revealed eight targets that were actively involved in developmental processes.Of the predicted miRNA-mRNA pairs, six targets were then randomly selected and experimentally validated by dual luciferase reporter assay, where three pairs were proved with potential targeting activity.Overall, to search for conserved miRNAs potentially involved in early development of M.japonicus, we combined in silico and experimental methods, which can be applied in other organisms as well.Our data implied important roles of miRNAs in the early embryonic development and also suggested the presence of complex miRNA-mRNA functional networks in M.japonicus.
Keyword: microRNAs (miRNAs); identification; Marsupenaeus japonicus; embryo; transcriptome
1 INTRODUCTION
MicroRNAs (miRNA) is a type of endogenous and short non-coding RNAs, generally 18-24 nt in length, and it can spatiotemporally regulate the expression of target genes at post-transcriptional level in most organisms (Bartel, 2004; Carthew and Sontheimer, 2009).Like the protein-encoding genes,miRNA-encoding genes also need a stepwise scenario for their maturation.Primary miRNAs (primiRNAs) are first transcribed from genomic loci with 5′ caps and 3′ poly (A) tails and then processed into precursor miRNAs (pre-miRNAs) with partially complemented hairpin structure (Denli et al., 2004;Zeng and Cullen, 2005).Pre-miRNAs are then exported into cytoplasm and subsequently processed into miRNA duplexes by Dicer, and then mature miRNAs by helicase (Bernstein et al., 2001; Lund et al., 2004).The mature miRNAs within RNAinduced silencing complex (RISC) guide the silencing of target genes by direct degradation or translational inhibition (Ambros, 2004).Recently,increasing number of miRNAs were shown to be indispensable for cell fate determination, cell cycle regulation, tumorigenesis, and organ morphogenesis in animals.For example, murine miR-34 and miR-499 were found to contain similar seed sequence and share similar target genes.Later, they were reported to induce cell cycle arrest by transactivatingp53and inhibiting E2F signal pathway(Lizé et al., 2011).Zebrafish embryos with mutantdicergene showed abnormal morphogenesis during gastrulation, brain formation, somitogenesis and heart development.Moreover, defect in brain development could be rescued by introducing miR-430 (Giraldez et al., 2005; Wienholds et al., 2005a).Taken together, it is believed that miRNAs play important roles in biological activities and developmental processes.
A huge number of miRNAs have been identified and characterized in animals and plants, such as flyDrosophilamelanogaster(Lai et al., 2003), zebrafishDaniorerio(Kloosterman et al., 2006), thale cressArabidopsisthaliana(Adai et al., 2005) and maizeZeamays(Zhang et al., 2006a).Today, both experimental methods and in-silico predictions are used to discover novel miRNAs.Experimental methods are time-consuming and inefficient, especially for low abundant miRNAs.Genomic and expressed sequence tag (EST) datasets have become important sources to search for miRNAs by in-silico prediction using known miRNAs as references.However, only conserved miRNAs across different organisms could be identified in this process (Wan et al., 2012).High-throughput sequencing made the direct construction of small RNA libraries come true and lots of miRNAs have been found, especially those in low abundance and specific species.
In aquatic organisms, a plenty of miRNAs have been identified by experimental methods or bioinformatic analysis.Dissecting miRNA profiles has been reported in different fish species(Ramachandra et al., 2008; Brzuzan et al., 2010; Chi et al., 2011; Xie et al., 2011; Barozai, 2012; Xu et al., 2012), crustaceans (Li et al., 2013; He et al.,2015; Ünlü et al., 2015; Lv et al., 2016) and mollusks (Yu et al., 2012; Martín-Gómez et al.,2014; Zhou et al., 2014; Kenny et al., 2015).For aquatic organisms, further studies displayed that miRNAs exerted a variety of cellular and physiological functions ranging from regulation of immunity(Chen et al., 2014; Sha et al., 2014; Zhang et al.,2014), organogenesis (Kapsimali et al., 2007;Ramachandran et al., 2010) and reproduction(Bizuayehu et al., 2012; Abramov et al., 2013; Xiao et al., 2014) to lipid and ion homeostasis (Flynt et al., 2009; Yan et al., 2012; Ordas et al., 2013).In shrimps, miRNAs were characterized in Pacific white shrimpLitopenaeusvannamei(Guo et al.,2018; Wang et al., 2019, 2020; Millard et al., 2021),kuruma shrimpMarsupenaeusjaponicus(Zhu et al.,2016; Zheng et al., 2018), black tiger shrimpPenaeusmonodon(Li et al., 2020; Kanoksinwuttipong et al., 2022), Japanese freshwater prawnMacrobrachiumnipponense(Ou et al., 2022a, b),and Chinese shrimpFenneropenaeuschinesis(Li et al., 2017b, 2019; He et al., 2019).However, a majority of the above reports were devoted to functional study of miRNAs in viral infection and other stresses.Therefore, we hypothesized that miRNAs may also play roles in regulating developmental process in shrimps.In the present study, based on the mRNA transcriptomic dataset from the gastrula embryos ofM.japonicus,conserved miRNA candidates were predicted by bioinformatics approach and then experimentally validated.Subsequently, potential miRNA-mRNA interactions were further investigated to uncover development-related target genes.To our best knowledge, this study is the first report of transcriptome-wide identification and characterization of miRNAs from early embryos ofM.japonicus,which provides valuable information for future work on the function of shrimp miRNAs in embryonic development.
2 MATERIAL AND METHOD
2.1 Shrimp embryo and sampling
After spawning, fertilized eggs ofM.japonicuswere collected from a local shrimp breeding farm(Jiaonan, Qingdao, China), transferred to laboratory immediately and maintained in aerated seawater(26-28 °C).Then the embryos at gastrula and nauplius stage were sampled at 6-7 h and 10-11 h post spawning, respectively.After suspended in Trizol reagent (Transgen), the samples were rapidly freezed and stored at -80 °C for RNA isolation.
2.2 Transcriptome dataset of gastrula embryos of M.japonicus
The whole transcriptome dataset was derived from our previous study in our laboratory (Li et al.,2017a).In briefly, the total RNA of gastrula embryos was isolated using the Trizol reagent(Transgen).After treated with RNase-free DNase I(TaKaRa, Japan) in the presence of RNase inhibitor(TaKaRa, Japan) according to manufacture protocol,the total RNA was then subjected to reverse transcription and cDNA library was constructed.The Illumina paired-end sequencing of mRNA transcriptome was carried out by Novogene Company (Beijing, China).After removing the reads with low quality and adapter sequences, all the clean reads were assembled to transcripts using Trinity software (Grabherr et al., 2011; Haas et al., 2013).
2.3 Cell and cell culture
The mammalian Neuro-2a cell, purchased from ATCC cell bank, was a fast-growing mouse neuroblastoma cell line.Neuro-2a cells were maintained in Dulbecco’s modified Eagle’s medium(DMEM, Life Technologies) supplemented with 10% bovine calf serum (BCS, Life Technologies),and cultured at 37 °C in a 5% CO2incubator.
2.4 Transcriptome-wide identification of mjamiRNAs in gastrula embryo
To uncover the conserved miRNA candidates from shrimp embryo, unique miRNA references including 4 053 non-redundant known microRNAs deposited in miRBase (Release 20, June 2013; http://www.mirbase.org) from 19 species was used as query sequences to blast against the 67 183 transcripts from the transcriptome dataset ofM.japonicusembryos (Li et al., 2017a).The alignment search was carried out through local BLASTN program(Zhang et al., 2000).The sequences with less than 3 nt mismatch with query miRNA and E-value lower than 0.5 or score >30, allowing for a minimum of 18 nucleotides in length were manually chosen for the subsequent analysis.All the predicted mature miRNA sequences along with their 200-bp upstream and 200-bp downstream flanking sequences were selected from the transcripts and were subjected to RNAfold web server (http://rna.tbi.univie.ac.at/cgibin/RNAfold.cgi) to generate the secondary structure with default parameters (Hofacker, 2003).The harpin structures according to the following strict criteria could be considered as the precursor miRNAs(pre-miRNA) inM.japonicusembryos: the RNA sequence did not contain more than 3 nt mismatches with the query homology and could be folded into stem-loop hairpin secondary structure; mature miRNA sequence was not located in the terminal loop of the hairpin structure but the same arm of the stem-loop hairpin structure; mismatches between miRNA and miRNA* was less than 6 nt; the folding hairpin structure had higher minimal folding free energy index (MFEI) than other non-miRNAs; a potential miRNA sequence could not contain large loops or breaks in microRNA: microRNA* duplex(Yin et al., 2008; Wang et al., 2012a).
2.5 Conservation analysis
The 26 714 miRNAs from metazoon were collected from miRBase to create the local miRNAs dataset.All identified miRNA candidates derived fromM.japonicusembryo were searched against the local miRNA dataset using BLASTN program with E-value cutoff of 10-2.The conservation was demonstrated by the distribution of mja-miRNAs homologue across different species (Apismellifera,Acyrthosiphonpisum,Drosophilamelanogaster,Nasoniavitripennis).Also, pre-miRNA of mja-miR-14 was chosen and its conservation with its orthologue in other species was analyzed using WebLogo, a sequence logo generator (Crooks et al.,2004).
2.6 Expression analysis of mja-miRNAs in gastrula and napulius stages by RT-qPCR
To experimentally validate the existence of identified mja-miRNAs, RT-qPCR was performed in this study to detect their expression.Furthermore, to gain insight into potential roles of mja-miRNA in regulating shrimp developmental process, differentially expressed mja-miRNAs (DEM) in two representatively developmental stages, gastrula and nauplius, were investigated.The total RNAs were isolated from gastrula and nauplius stages using mentioned methods above.After treating with RNase-free DNase I to remove possible genomic DNA contamination, two micrograms of total RNAs were used for miRNA RT-PCR reaction using SYBR PrimeScriptTMmiRNA RT-PCR Kit (TaKaRa,China).In brief, all the miRNAs in the total RNAs were first ployadenylated by Poly (A) polymerase and then reversed into miRNA cDNAs by PrimeScript RTase and each miRNA cDNA was tailed a uni-miR qPCR primer binding site by universal adaptor primer.miRNA RT-qPCR was performed on a Light Cycler Roche 480 Real-Time PCR System using SYBR Premix E×TaqII kit.The sequences of qRT-PCR primers were listed in Supplementary Table S1.The relative expression level of each miRNA transcript was automatically calculated as 2-ΔΔCtusing actin as an internal control.The Cp value (cycle threshold) was obtained using Light Cycler Roche 480 software.All the RT-PCR amplifications were repeated at least three times.One-way ANOVA followed by Tukey’s post-hoc test was carried out for statistical and significance analysis with software SPSS 20.0 (SPSS, IL, USA).
2.7 Prediction of target genes of mja-miRNAs
Mature miRNAs bind to mRNA to form a miRNA: mRNA duplex in perfect or near-perfect complementation, giving rise to gene expression regulation at post-transcription level.So it is very important to get insight into miRNA target genes to understand the regulation mechanism of miRNAs in the embryonic development ofM.japonicus.Today,bioinformatics approach has been an efficient strategy to predict mass target genes in comparison with experimental methods.In this study, all unigenes derived from de novo assembled transcriptome were blasted against the following protein database according to the order of NR,Swissport and KEGG GENES database to form the dataset of protein-encoding unigene sequences.As the existence of minus strands in transcriptome, all the minus strands were abstracted from this unigene dataset using perl scripts and converted into their reverse complementary sequences.Finally, a proteinencoding unigene dataset, only containing the plus strands, was built and used as a reference for the prediction of mja-miRNAs target genes.A widely used computational algorithm of miRanda was employed to predict the target genes (John et al.,2004).The criteria were used in the algorithm according to our previous study by Pu et al.(2018).In brief, to minimize the false-positives to a large extent, the following parameters were set more strictly: (1)S, the sum of single-residue-pair match scores over the alignment, ≥160; (2) ΔG, the free energy of duplex formation, <-25 kcal /mol; (3) strict,requiring strict alignment in the seed region (offset positions 2-8); (4) number of mismatches was not beyond three; (5) mismatches occurred between the 2ndand the 12thposition of the 5′ end were not more than one; (6) one gap at most was allowed,but not in seed region and cleavage sites.All the sequences meeting the above criteria were manually chosen and considered as the final mjamiRNA targets.
2.8 GO and KEGG analysis of the predicted target genes
To further understand the functions and metabolic pathway of target genes, analysis based on gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) database were performed using Blast2go suit (Ashburner et al., 2000;Kanehisa and Goto, 2000; Conesa et al., 2005; Götz et al., 2008).First of all, the target genes were screened for non-redundant protein database in NCBI with E-value cutoff of 1e-5.Then the function category of the best BLAST hit was assigned using the function for mapping of GO terms with default parameters.Finally, GO annotation for target genes and pathway analysis were carried out by KEGG database.
2.9 Experimental validation of the predicted target genes
To get more insight into the roles of mjamiRNAs in the embryonic development of shrimps,the targeting activity of mja-miRNAs to the predicted genes involved in developmental process was experimentally validated by dual luciferase reporter gene assay system (Galen Biopharm International Co.Ltd., Beijing, China).Three mjamiRNAs and their corresponding target genes were selected for validating their interactions: mja-miR-6489-3p and its target of comp41742; mja-miR-3885 and its three targets of comp36129,comp45393, and comp42770; mja-miR-2491 and its two targets of comp41305 and comp36129.Three methylated single-strand miRNA mimics of mjamiR-6489-3p (5′-CGACGGAAAGGUGUCCAAGCUGG-3′), mja-miR-3885 (5′-AAGGCGGCGGCGGCGGCC-3′),and mja-miR-2491 (5′-CAACAACAGCAGCAGCAA-3′), as well as one negative miRNA mimic control (5′-UCACAACCUCCUAGAAAGAGUAGA-3′) were synthesized by Shanghai GenePharma Co.Ltd.(China).
To prepare the targeting site fragments of the six target genes mentioned above, only 52 bp in length flanking the predicted targeting site was chosen and used to design a pair of complementary singlestrand oligonucleotides with an addition of XhoI or NotI sites at both ends (59 bp in length each), which were artificially synthesized by BGI company(Shenzhen, China), respectively.These primers were then subjected to annealing reaction after 5′-end addition of phosphate and a double-strand insertion unit for each target gene was obtained.The annealing reaction was performed in 20-μL total volume consisting of 8-μL oligonucleotide (0.1 nmol/μL) for each strand, 0.5-μL ATP-Na2(10 mg/mL), 0.4-μL T4 polynucleotide kinase (10 U/μL), 2-μL 10×T4 PNK buffer and 1.1-μL ddH2O and then incubated at 37 °C for 30 min, followed by 95 °C for 5 min and cooled to room temperature.After that, each doublestrand oligonucleotide obtained was directionally inserted into a reporter plasmid of psiCHECK-2, a kind gift from Dr.Haolin XU (College of Biological Sciences, Chinese University of Hong Kong).The ligation reaction mix in 15-μL total volume included 10 μL of the obtained 59-bp double-strand oligonucleotide, 0.7-μL linear psiCHECK-2 plasmid DNA (0.5 μg/μL), 1-μL T4 ligase (350 U/μL), 1.5-μL 10×T4 ligase buffer, and 1.8-μL ddH2O.In addition,for each of the six target genes tested, a mutant double-strand 59-bp oligonucleotide fragment with mutations introduced in the complementary sequence against the seed region of mja-miRNAs was also prepared in the same way as mentioned above.All of the synthesized wild-type and mutant target site fragments will be provided upon request.Next, the dual luciferase reporter gene assay was performed to validate the mja-miRNA targets.
2.10 Dual luciferase reporter gene assay
For dual luciferase reporter gene assay, all the stock solutions of fourteen psiCHECK-2 recombinant plasmids containing wild-type (WT) or mutant targeting sites, four synthesized single-strand mjamiRNA mimics and one synthesized negative control mimic were prepared in RNase-free water and adjusted to a final concentration of 25 μmol/L.One day prior to transfection, Neuro-2a cells were seeded into each well of a 24-well culture plate.Approximately 60%-70% confluence was reached at the time of transfection.To validate the targeting between mja-miRNAs and their target genes tested,varied psiCHECK-2 recombinant plasmid DNAs containing wild-type or mutant target gene regions were co-transformed with their corresponding miRNA mimic or negative control mimic into the Neuro-2a cells using Lipofectamine 2000 transfection reagent (Invitrogen) following the product manual.Four groups were tested: 1) Negative mimic control(1.6 μL)+psiCHECK-2-WT (4 μL); 2) miRNA mimic(1.6 μL)+psiCHECK-2-WT (4 μL); 3) Negative mimic control (1.6 μL)+psiCHECK-2-mutant (4 μL); 4)miRNA mimic (1.6 μL)+psiCHECK-2-mutant (4 μL).For each group, all volumes were multiplied by 3.5 to account for the triplicate samples and liquid loss during pipetting.
At 48-h post-transfection, the old medium in each well was removed and the cells were rinsed with PBS buffer (Han et al., 2013) twice.Then the activities of firefly luciferase andRenillaluciferase were measured using dual luciferase reporter gene assay kit.In brief, after cell lysis and centrifugation,20 μL of cell lysate supernatant was added into a 1.5-mL centrifuge tube containing 100 μL of luciferase assay reagent, mixed quickly and the firefly luciferase activity was measured immediately using luminometer (GloMax, Promega).Then 100 μL of stop reagent was added and briefly mixed to recordRenillaluciferase activity.As theRenillaluciferase gene was set as reporter gene and firefly luciferase gene was set as inner reference in the plasmid map of psiCHECK-2, the ratios ofRenillaluciferase activity to firefly luciferase activity were calculated for each sample and used to detect the targeting activity between mja-miRNAs and target genes.The SPSS software was used for data statistical analysis.
3 RESULT
3.1 Transcriptome-wide survey of embryodominant mja-miRNAs
As shown in Fig.1, a total of 21 conserved mjamiRNAs were identified by homologous searching against the transcript sequences, comprising of 19 families (miR-14, 44, 242, 287, 317, 927, 966, 969,1014, 1175, 2491, 2731, 2774, 2788, 3338, 3885,4961, 6489, and 6493).Five families (miR-317,966, 2788, 6489, and 6493) have already been reported by other works inM.japonicus(Ruan et al., 2011; Huang et al., 2012; Zheng et al., 2018; He et al., 2022), which indicated the reliability of our methods used here.For the other 14 families and their members, they were reported for the first time inM.japonicus.Among the 21 embryo-predominant miRNAs, 13 (61.90%) of them were located in the 3′ arm of hairpin structures and the remaining 8(38.10%) were in the 5′ arm of hairpin.The length of mature miRNAs ranged from 18 to 24 nt with the average length of 20.48 nt.The mature miRNAs with the length of 20 nt and 21 nt were the majority source of the total miRNAs, accounting for 19.05%and 28.57%, respectively (Fig.2a).

Fig.1 Sequences and harpin structures of the identified embryo-predominant mja-miRNAs from transcriptomic dataset
It has been reported that, the four nucleotides of A, U, G, and C distribute unevenly in the precursors, and no matter in plant or animals, the nucleotide uracil is the most predominant base presented in mature miRNAs and pre-miRNAs(Wang et al., 2012b).In this study, the four nucleotides accounted for 23.28%±7.44% (A),25.62%±9.64% (U), 24.52%±9.09% (G), and 26.59%±7.46% (C), respectively (Fig.2c).Except for C content, the average of U content was higher than those of A and G without significant difference(t-test,P>0.05).As G and C could form three hydrogen bonds, more GC content the hairpin contains, more stable the secondary structure is.Then the AU and GC contents in precursor were also calculated.It was found that AU content(48.99%±15.38%) was lower than GC content(51.01%±15.38%), which is responsible for the structure stability (Fig.2b).The ratios of A/U and C/G were also investigated and the values of both ratios were 0.92 and 1.03, respectively, which suggested that more U and C existed in miRNA precursors.MEFI is another important parameter for miRNA secondary structures and it is always higher than other types of RNA.In this study, the MEFI ranged from 0.62 to 1.06 with the average of 0.75±0.13,which is in accordance with previous studies (Zhang et al., 2006b).The statistic results for each parameter were listed in Table 1.
miRNA families often contain one member.However, we found in the present study that both family 6489 (mja-miR-6489-5p and mja-miR-6489-3p) and 6493 (mja-miR-6493-5p and mja-miR-6493-3p) had two members.Moreover, these four miRNAs were clustered into the same transcript or target gene (comp41409_c0), suggesting that they might share a common promoter and be cotranscribed from the genome (Fig.2d).Recently, as differentially expressed miRNAs responded to virus infection, miR-6489-3p and miR-6493-5p were both identified in intestinal tissue ofM.japonicus(He et al., 2022), further supporting our proposed model of co-transcription.
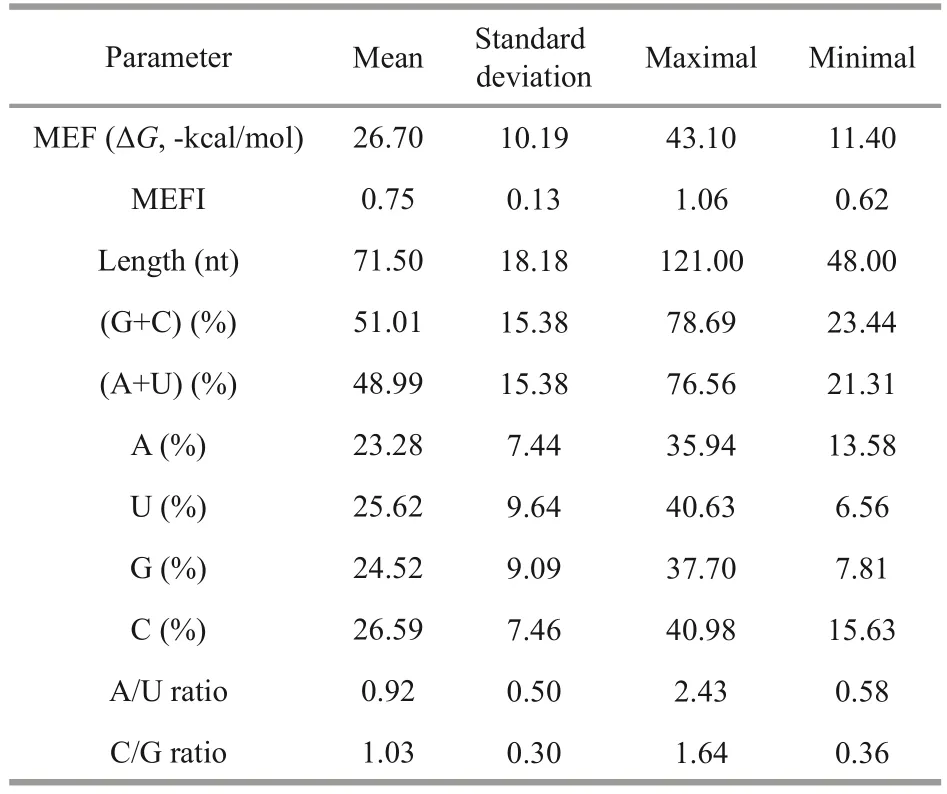
Table 1 Statistical results of the characterized parameters of mja-miRNA precursors
3.2 Conservation analysis
Numerous of studies have reported that miRNAs sequences are highly conserved among different species (Wienholds and Plasterk, 2005b).The mjamiRNAs identified in this study were searched against all mature miRNAs in metazoon (E-value<0.01)and found that 17 mja-miRNAs showed conservation in other species, of which two mja-miRNAs (mjamiR-287 and mja-miR-14) were highly conserved across more than ten species (Fig.3a).
In addition to the mature miRNA sequences, the pre-miRNA sequences are also reported to be conserved among different organisms.Herein, one representative pre-miRNA of mja-miR-14 was selected to perform the conservation analysis(Fig.3b).As expected, the precursor sequences were highly conserved too, demonstrating that mjamiRNAs were conserved not only in mature sequences, but also in precursor sequences.

Fig.2 Characterization of mja-miRNAs in M.japonicus embryo
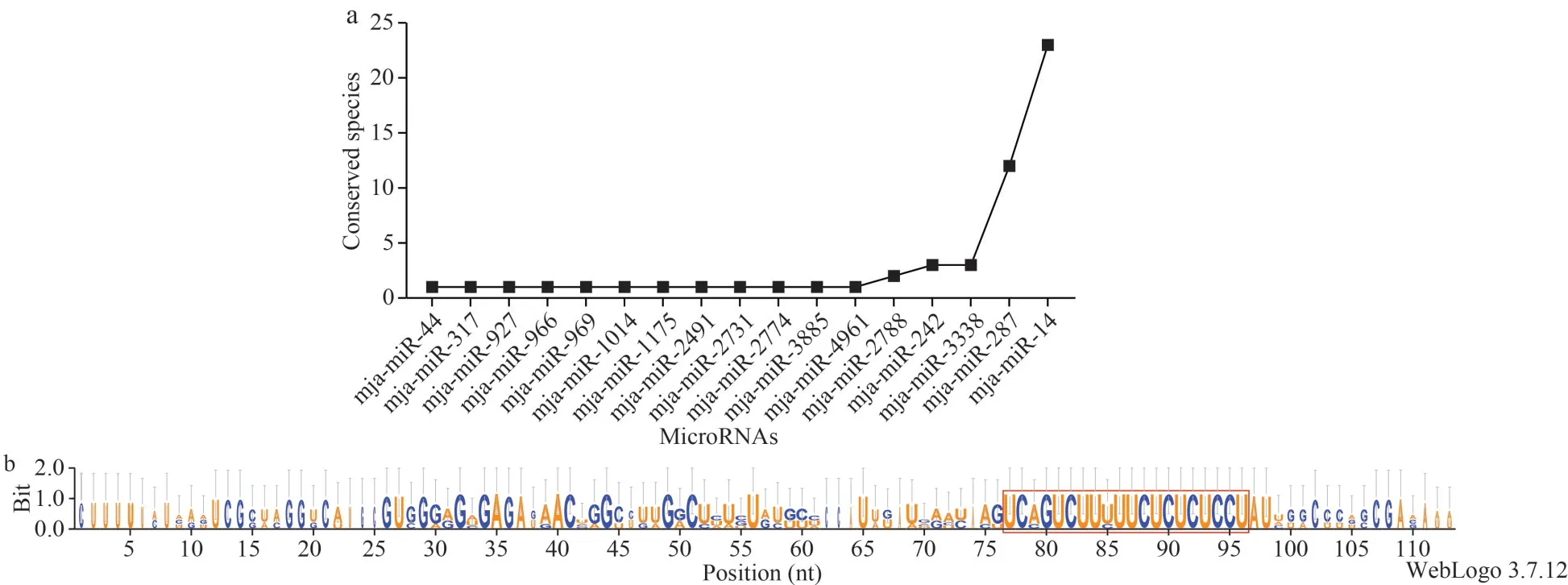
Fig.3 Conservation analysis of the identified mja-miRNAs and the representative pre-miRNA of mja-miR-14
3.3 Validation of predicted mja-miRNAs by RTqPCR and identification of differentially expressed miRNAs between gastrula and napulius stages
To verify the existence of predicted mjamiRNAs, RT-qPCR was performed to detect their expression.Of the identified 21 mja-miRNAs,fifteen were successfully amplified from gastrula stage (Fig.4a).As shown in Fig.4b, verified mjamiRNAs in gastrula stage varied dramatically, and five mja-miRNAs including miR-3885, 6493-5p,6489-5p, 6489-3p, and 2774 displayed relatively high expression level compared with others.Subsequently, we asked whether these existed mjamiRNAs present dynamic changes along with different developmental stages.Expression profiles of mja-miRNA in gastrula and nauplius were compared, and as shown in Fig.4c, most of mjamiRNAs displayed different expression level in two stages.Compared with those in gastrula stage, seven and four miRNAs were significant upregulated or downregulated in nauplius, respectively.However,other four mja-miRNAs expressed consistently during shrimp development.This data indicated each developmental stage harbored distinct miRNA profile and these differentially expressed mjamiRNAs inferred the involvement of miRNAs in regulating development in shrimps.
3.4 Functional characterization of target genes for embryo-dominant mja-miRNAs
Identification of the target mRNA of each mjamiRNAs could provide clues for understanding the roles of miRNAs in regulation of development.Even though some predicted mja-miRNAs were failed to be detected by RT-qPCR, we can not exclude the possibility that it was caused by their low abundance.Hence, all the identified mja-miRNAs were formed as custom miRNA dataset in this study.And the unigenes from gastrula transcriptome were regarded as the custom target dataset.A total of 120 mRNA sequences were predicted from the gastrula transcriptome ofM.japonicasas targets of 12 mja-miRNAs.The number of predicted targets varied for different mja-miRNAs.Most of the targeting genes were assigned to mja-3885 (33 targets) and mja-2491 (30 targets), followed by mjamiRNA-966, mja-miRNA-14, and mja-miRNA-317(Supplementary Table S2).And here we did not observe the positive correlation between miRNA expression and targeting gene number.
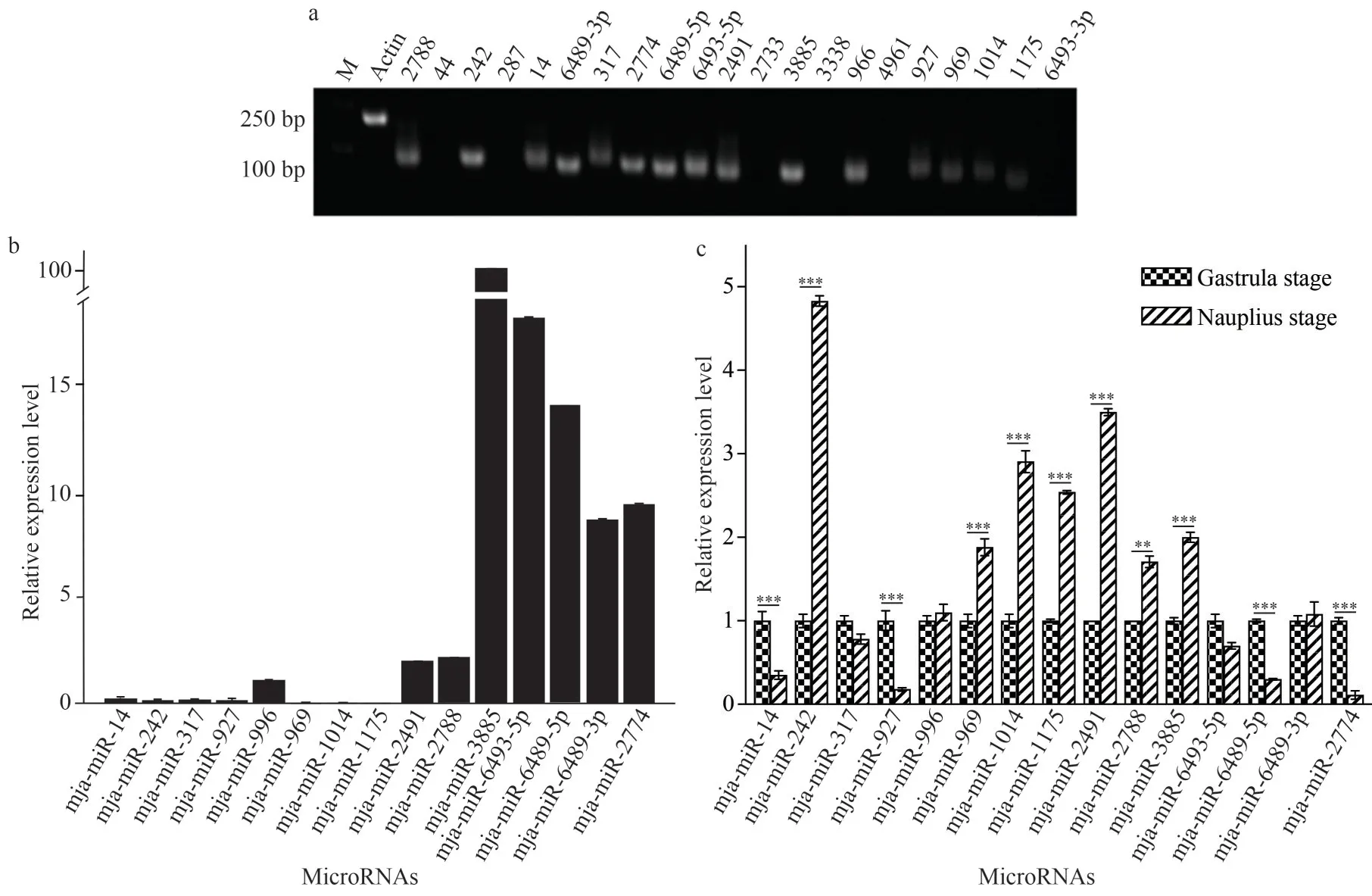
Fig.4 RT-qPCR validation of mja-miRNAs and differentially expressed miRNAs (DEM) analysis
Gene silencing mediated by miRNAs functions via base-pairing with complementary sequences within mRNA molecules.This function may occur either via mRNA degradation or protein translational inhibition (Ambros, 2004).For mRNA degradation,miRNAs are guided to a complementary site by RISC, which then Ago2 cleaves the mRNA followed by the direct mRNA degradation.This process will lead to the reduction of mRNA abundance.RNA abundance of target genes is an important index for understanding miRNA: mRNA interaction and its regulation in networks.As for high-throughput RNA sequencing methods, such as Illumina sequence data, the reads per kilobase per million reads(RPKM) measure was widely used to examine RNA abundance (Mortazavi et al., 2008).Here, RPKM values of all target genes were calculated and used as a reference for mRNA abundance analysis.As shown in Fig.5a, the values of 85 (70.83%) target genes were less than 10 and even there were 9(7.5%) targets with the RPKM values less than 1.0,which also suggested the reliability of target prediction in this study.Meanwhile, 35 (29.17%)target genes had the RPKM values more than 10.0 and even some were shown with extremely high values.There are two possible explanations for these highly expressed mRNAs.One is that RPKM values derived from high throughput sequencing are not so accurate that the divergences occur for some gene transcripts.Another possibility is that gene silencing for these targets are not achieved by degrading mRNA, but rather by disturbing efficient translation into proteins.
To get more insight into the function of miRNA targets and networks between miRNAs and targets,all the predicted target genes were subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database for analysis.GO terms were categorized into three classes of cellular component, molecular function and biological process (Ashburner et al., 2000).The results showed that, at GO term level 2, all target genes were assigned to 11 categories at cellular component level, 10 categories at molecular functions level and 18 categories at biological process level.The distribution of assigned GO terms was also produced by WEGO output to visually show gene function.As shown in Fig.5b, 50.83% miRNA targets were located in cell and cell part, followed by those located in organelle (25%) and macromolecular complex (17.5%).At molecular function level,strikingly more targets were assigned to binding(57.5%) and catalytic activity (29.17%).Cellular process (49.17%), metabolic process (39.17%),biological regulation (27.5%) and pigmentation(26.67%) were four dominant biological processes.Subsequently, KEGG pathway analysis was also performed for pathway enrichment.The results show that 19 targets were involved in 31 metabolic pathways (Supplementary Table S2), indicating that one target participated in several pathways or several targets shared one pathway.The major metabolic pathways were: RNA degradation, Wnt signaling pathway, cell cycle and mineral absorption and so on, all were crucial events in shrimp embryonic development.
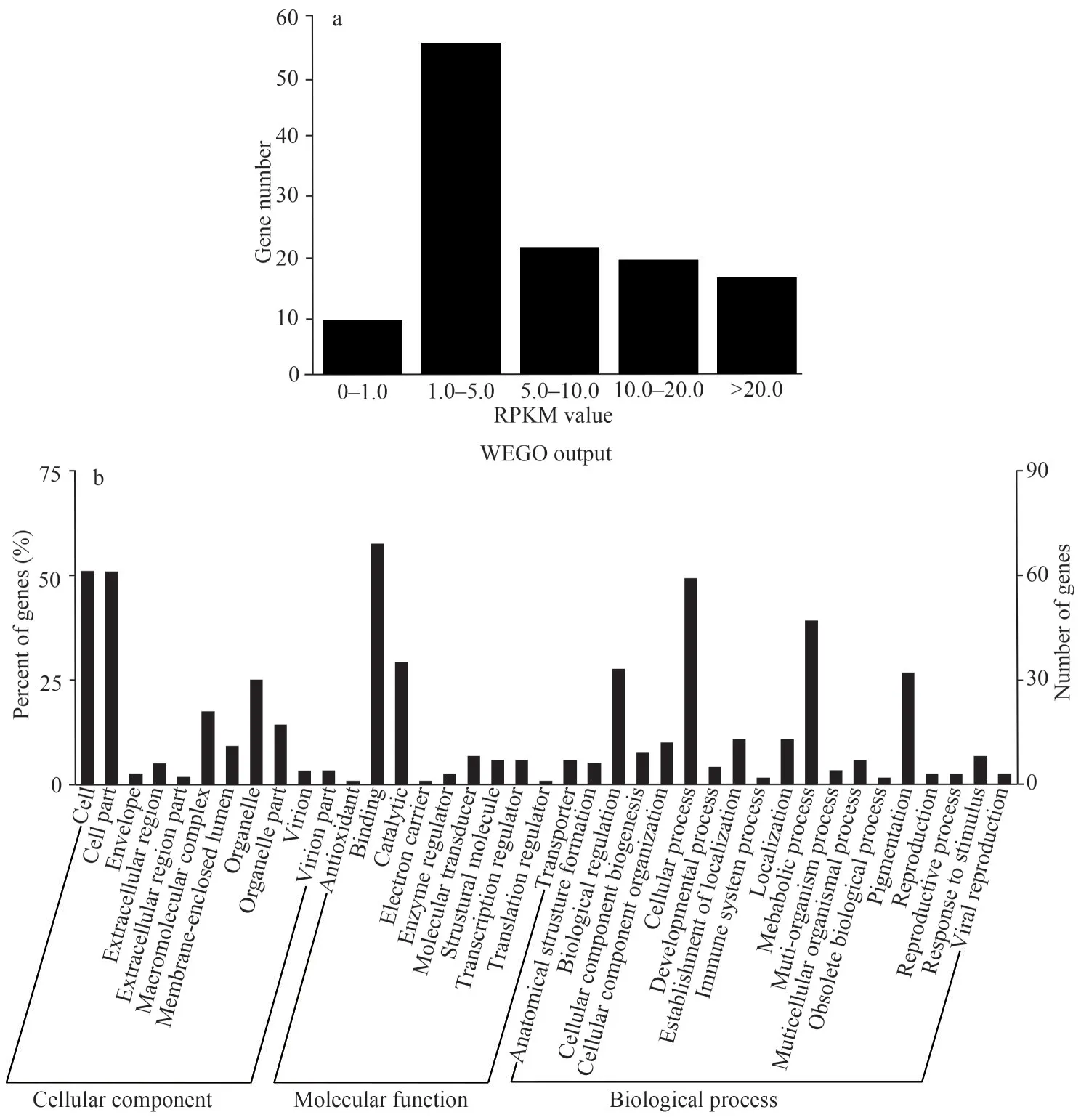
Fig.5 The distribution of the reads per kilobase per million reads (RPKM) values of 120 target genes predicted from the gastrula transctiptome of M.japonicus (a); gene ontology categories and distribution of embryo-predominant mjamiRNAs target genes (b)
3.5 Experimental validation of miRNA-mRNA interactions involved in embryonic development
Furthermore, in the process of target annotation,some genes were found to be closely related to developmental activities, termed as developmentrelated target genes.A total of eight genes targeted by five mja-miRNAs (miR-2731, 2491, 3885, 4961,and 6489-3p in Table 2) were collected from the target pool to form this cluster, which potentially played key roles in regulating embryonic development ofM.japonicas.
As we thought, this cluster of target genes is critical in embryonic development.To facilitate our understanding of miRNA-mediated regulation, we verified miRNA-mRNA interactions experimentally.Among the five mja-miRNAs and their corresponding target genes, targeting activity of three PCRconfirmed mja-miRNAs (miR-2491, 3885, and 6489-3p) to their six target candidates was investigated in Neuro-2a cells by dual luciferase reporter gene assay.As shown in Fig.6, downregulation of target genes by miRNA mimic was found in three groups: comp41305 targeted by mjamiR-2491, comp36129 targeted by mja-miR-2491,and comp45393 targeted by mja-miR-3885.However,for the remaining three experimental groups, it was failed to detect obvious targeting activity (data not shown), indicating that prediction data based on computational algorithm need to be experimentally validated to avoid potential false-positive results.Due to the absence of immortalized shrimp cell line,our data obtained by using mammalian cell line asplatform provided an alternative method for validating miRNA-mRNA interactions in shrimps.

Table 2 The list of eight development-related target genes
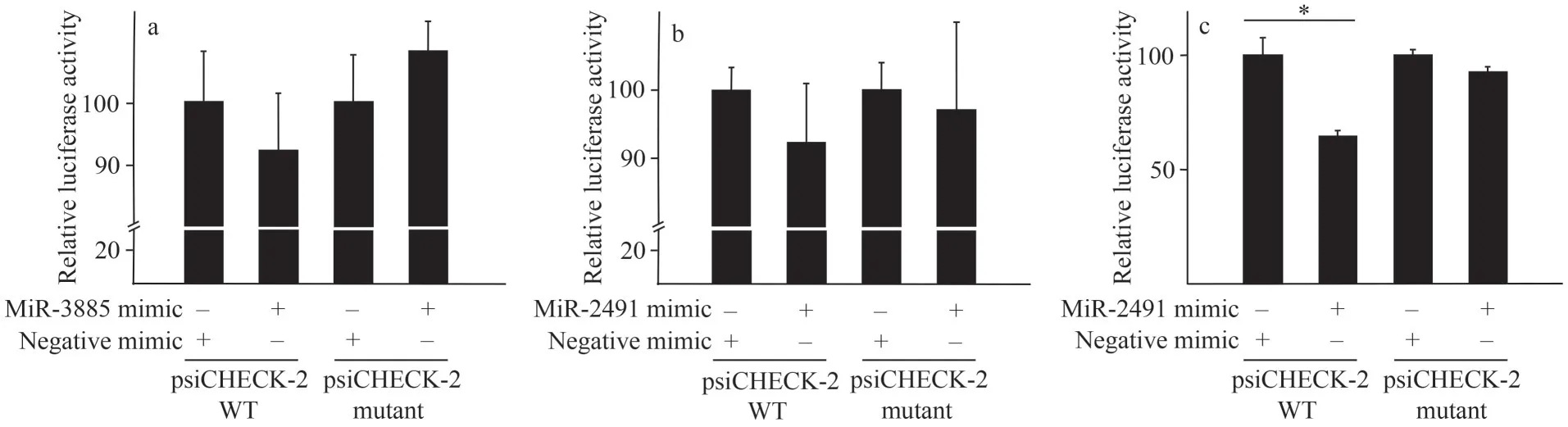
Fig.6 Experimental validation of the targeting of 2 mja-miRNAs to their corresponding target genes in Neuro-2a cells using dual luciferase reporter assay
4 DISCUSSION AND CONCLUSION
In contrast to plant miRNAs, the length of the majority of animal pre-miRNAs was more consistent,typically 70-80 nt (Yin et al., 2008).Similar results were found in this study, that is, the length of all identified mja-miRNA precursors ranged from 48 to 121 nt with the average length of 71.50±18.18 nt,and the precursors containing 55-90 nt accounted for 85.51%.Of those nineteen families, only five families inM.japonicushave been reported.One possible explanation is that miRNAs in shrimps are tightly regulated and express in specific tempo-spatial manner.Investigation of miRNAs in single tissue or development stage could not represent the global expression profile and shall miss a lot of miRNAs expressed in other events.Meanwhile, except for the ones reported in this study, we expected that there must be more mja-miRNAs to be identified in future, which can regulate shrimp development as well.One class of such mja-miRNAs are those with very low expression level.And another class of mjamiRNAs are those un-conserved ones.It is a pity that, for both of them, it is hard to be discovered by our methods used in the present study and complementary techniques are needed to be performed,such as high-throughput miRNAs sequencing.
For RT-qPCR amplifying failure of six mjamiRNAs (mja-miR-44, 287, 2733, 3338, 4961, and 6493-3p) in both gastrula and nauplius stages, the low abundance or improper primers might account for it.Intriguingly, mja-miR-6489-5p, 6489-3p, and 6493-5p have been found to cluster into one transcript (comp41409_c0), thus their expression was detected in both gastrula and nauplius, which further supported our hypothesis these mja-miRNA genes clustering into the same transcript cotranscribed simultaneously.However, with the hypothesis we proposed here, it is hard to explain that mja-miR-6493-3p could not be detected in both stages using RT-PCR.Noteworthy, mja-miR-3885, a miRNA family also identified inTriboliumcastaneum,was expressed in extremely high level in both gastrula and nauplius, suggesting that mja-miR-3885 may play important roles in regulating shrimp developmental process.In addition, according to our results in this study, miRNAs expression is highly dynamic in terms of different miRNAs in the same developmental stage or the same miRNA in different stages.Given that miRNAs exert functions at posttranscriptional level, mRNA activity and abundance are precisely and tightly regulated, which might indicate that mja-miRNAs could actively participate in the developmental process ofM.japonicas.
To understand the role of mja-miRNAs in embryonic development, the target genes of all identified 21 mja-miRNAs were predicted using two sets of criteria, which were based on the complementary between miRNA and mRNA and the free energy of the duplexes.For most mja-miRNAs,more than one mRNA sequences were predicted as putative target genes, suggesting that our results were consistent with previous demonstration that one miRNA could have several target sites.Only two miRNAs, mja-miR-1175 and mja-miR-2778,just had one mRNA target predicted in this study.Of them, mja-miR-3885 had the largest number of putative target genes (34 target genes) including zinc finger protein, the most abundant proteins in eukaryotic genomes, and translation initiation factor,which appears to be involved in the regulation of gene transcription and mRNA translation (Miller et al., 1985; Laity et al., 2001).Moreover, cyclin-Gassociated kinase, protein phosphatase inhibitor,tyrosine-phosphorylation-regulated kinase which are responsible for signal activation, G-protein coupled octopamine receptor and type 1 dopamine receptor which are involved in signal transduction and amplification were also included.We assumed that multiple genes with diverse function targeted by miR-3885 resulted from its high expression level and thus, miR-3885 might played key roles in regulating embryonic development in shrimps.Another mja-miRNA targeting many genes was mjamiR-2491.Although the expression level was not as high as mja-miR-3885, nearly 4-fold up-regulation of its expression in nauplius took place compared with that in gastrula, suggesting mja-miR-2491 also played important roles in transition from gastrula to nauplius.The search of gene enrichment in gene ontology demonstrated many mja-miRNA target genes took part in significantly physiological activities and metabolic processes in embryonic development, further suggesting regulatory roles played by these miRNAs.Although a plenty of miRNA targets are predicted, undoubtedly false positives can still occur.So experimental validation is particularly needed via different methods.Since lacking of immortalized shrimp cell line and effective gene transfer technology in shrimps, an alternative in-vitro platform to validate shrimp miRNA-mRNA interaction can be employed, such as the dual luciferase reporter gene assay used in this study.Taken together, the current study presented a very complex regulation network woven by mja-miRNAs and their targets to govern the developmental process in shrimp.In addition, the study will even guide our future investigations of biological functions and mechanisms of mjamiRNAs in the embryonic development of shrimps and even in other crustaceans.
5 DATA AVAILABILITY STATEMENT
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
6 ACKNOWLEDGMENT
We thank Dr.Haolin XU (College of Biological Sciences, the Chinese University of Hong Kong) for kind gift of psiCHECK-2 plasmid and Sojeong JUN(Children’s Medical Center Research Institute and Department of Pediatrics, UT Southwestern Medical Center, USA) for language polishing.
杂志排行

Journal of Oceanology and Limnology的其它文章
- Contrasts of bimodal tropical instability waves (TIWs)-induced wind stress perturbations in the Pacific Ocean among observations, ocean models, and coupled climate models*
- Variability of the Pacific subtropical cells under global warming in CMIP6 models*
- Identification of thermal front dynamics in the northern Malacca Strait using ROMS 3D-model*
- Magmatic-tectonic response of the South China Craton to the Paleo-Pacific subduction during the Triassic: a new viewpoint based on Well NK-1*
- An improved positioning model of deep-seafloor datum point at large incidence angle*
- Microplastics in sediment of the Three Gorges Reservoir:abundance and characteristics under different environmental conditions*